Ghosh Manik C, Zhang De-Liang, Ollivierre Wade H, Noguchi Audrey, Springer Danielle A, Linehan W Marston, Rouault Tracey A
Molecular Medicine Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Murine Phenotyping Core, National Heart, Lung, and Blood Institute, and.
Blood. 2021 May 6;137(18):2509-2519. doi: 10.1182/blood.2020009138.
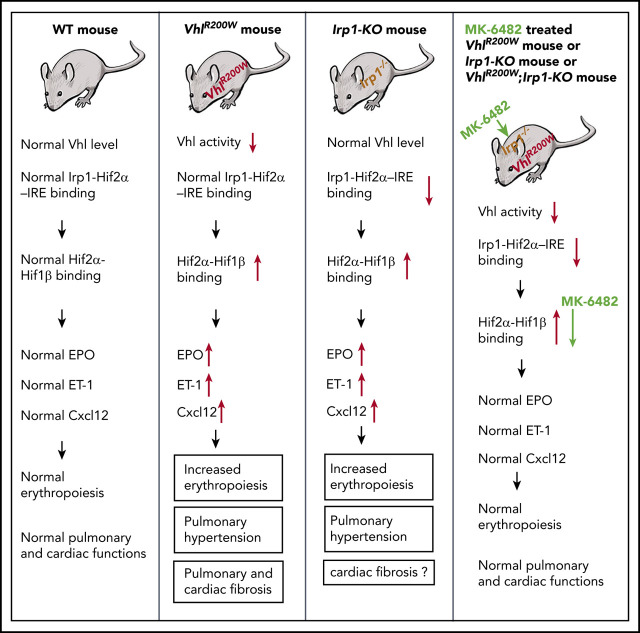
Polycythemia and pulmonary hypertension are 2 human diseases for which better therapies are needed. Upregulation of hypoxia-inducible factor-2α (HIF-2α) and its target genes, erythropoietin (EPO) and endothelin-1, causes polycythemia and pulmonary hypertension in patients with Chuvash polycythemia who are homozygous for the R200W mutation in the von Hippel Lindau (VHL) gene and in a murine mouse model of Chuvash polycythemia that bears the same homozygous VhlR200W mutation. Moreover, the aged VhlR200W mice developed pulmonary fibrosis, most likely due to the increased expression of Cxcl-12, another Hif-2α target. Patients with mutations in iron regulatory protein 1 (IRP1) also develop polycythemia, and Irp1-knockout (Irp1-KO) mice exhibit polycythemia, pulmonary hypertension, and cardiac fibrosis attributable to translational derepression of Hif-2α, and the resultant high expression of the Hif-2α targets EPO, endothelin-1, and Cxcl-12. In this study, we inactivated Hif-2α with the second-generation allosteric HIF-2α inhibitor MK-6482 in VhlR200W, Irp1-KO, and double-mutant VhlR200W;Irp1-KO mice. MK-6482 treatment decreased EPO production and reversed polycythemia in all 3 mouse models. Drug treatment also decreased right ventricular pressure and mitigated pulmonary hypertension in VhlR200W, Irp1-KO, and VhlR200W;Irp1-KO mice to near normal wild-type levels and normalized the movement of the cardiac interventricular septum in VhlR200Wmice. MK-6482 treatment reduced the increased expression of Cxcl-12, which, in association with CXCR4, mediates fibrocyte influx into the lungs, potentially causing pulmonary fibrosis. Our results suggest that oral intake of MK-6482 could represent a new approach to treatment of patients with polycythemia, pulmonary hypertension, pulmonary fibrosis, and complications caused by elevated expression of HIF-2α.
真性红细胞增多症和肺动脉高压是两种需要更好治疗方法的人类疾病。缺氧诱导因子-2α(HIF-2α)及其靶基因促红细胞生成素(EPO)和内皮素-1的上调,在携带von Hippel Lindau(VHL)基因R200W突变纯合子的楚瓦什真性红细胞增多症患者以及具有相同纯合VhlR200W突变的楚瓦什真性红细胞增多症小鼠模型中导致真性红细胞增多症和肺动脉高压。此外,年老的VhlR200W小鼠发生了肺纤维化,最可能是由于另一个Hif-2α靶标Cxcl-12的表达增加。铁调节蛋白1(IRP1)发生突变的患者也会发生真性红细胞增多症,Irp1基因敲除(Irp1-KO)小鼠表现出真性红细胞增多症、肺动脉高压和心脏纤维化,这归因于Hif-2α的翻译抑制解除以及Hif-2α靶标EPO、内皮素-1和Cxcl-12的高表达。在本研究中,我们使用第二代变构HIF-2α抑制剂MK-6482使VhlR200W、Irp1-KO和双突变VhlR200W;Irp1-KO小鼠中的Hif-2α失活。MK-6482治疗降低了所有3种小鼠模型中的EPO产生并逆转了真性红细胞增多症。药物治疗还降低了VhlR200W、Irp1-KO和VhlR200W;Irp1-KO小鼠的右心室压力并减轻了肺动脉高压至接近正常野生型水平,并使VhlR200W小鼠的心脏室间隔运动恢复正常。MK-6482治疗降低了Cxcl-12的增加表达,Cxcl-12与CXCR4一起介导纤维细胞流入肺部,可能导致肺纤维化。我们的结果表明,口服MK-6482可能代表一种治疗真性红细胞增多症、肺动脉高压、肺纤维化以及由HIF-2α表达升高引起的并发症患者的新方法。