Cole Casey A, Daigham Nourhan S, Liu Gaohua, Montelione Gaetano T, Valafar Homayoun
Department of Computer Science & Engineering, University of South Carolina, Columbia, South Carolina, United States of America.
Department of Molecular Biology and Biochemistry, and Department of Biochemistry, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Piscataway, New Jersey, United States of America.
PLoS Comput Biol. 2021 Feb 1;17(2):e1008060. doi: 10.1371/journal.pcbi.1008060. eCollection 2021 Feb.
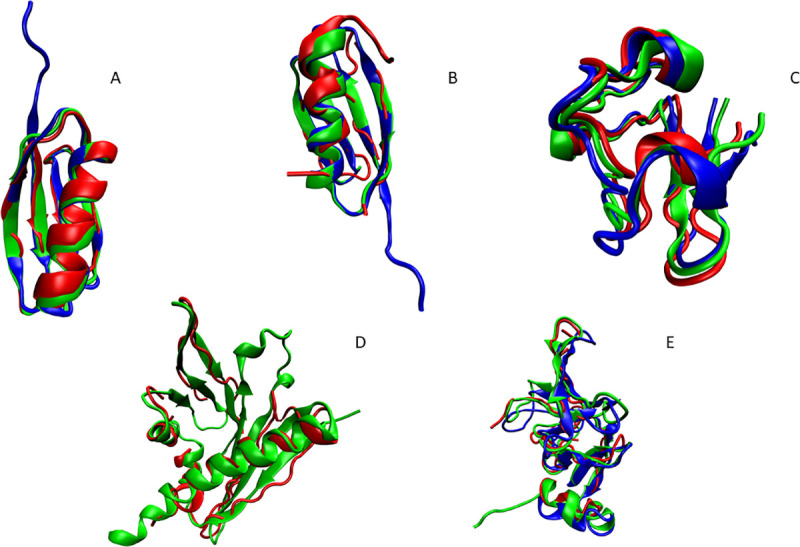
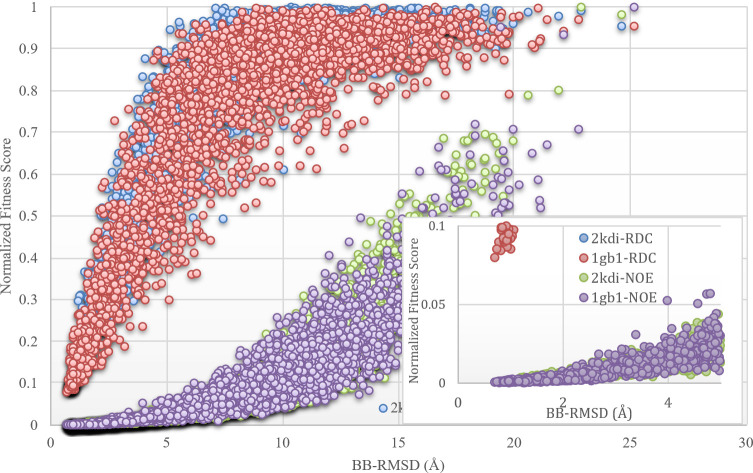
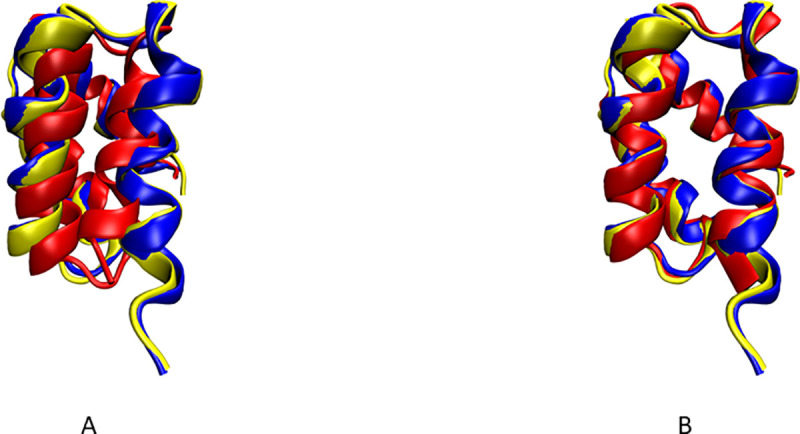
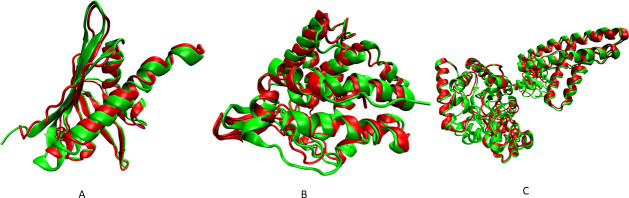
Nuclear Magnetic Resonance (NMR) spectroscopy is one of the three primary experimental means of characterizing macromolecular structures, including protein structures. Structure determination by solution NMR spectroscopy has traditionally relied heavily on distance restraints derived from nuclear Overhauser effect (NOE) measurements. While structure determination of proteins from NOE-based restraints is well understood and broadly used, structure determination from Residual Dipolar Couplings (RDCs) is relatively less well developed. Here, we describe the new features of the protein structure modeling program REDCRAFT and focus on the new Adaptive Decimation (AD) feature. The AD plays a critical role in improving the robustness of REDCRAFT to missing or noisy data, while allowing structure determination of larger proteins from less data. In this report we demonstrate the successful application of REDCRAFT in structure determination of proteins ranging in size from 50 to 145 residues using experimentally collected data, and of larger proteins (145 to 573 residues) using simulated RDC data. In both cases, REDCRAFT uses only RDC data that can be collected from perdeuterated proteins. Finally, we compare the accuracy of structure determination from RDCs alone with traditional NOE-based methods for the structurally novel PF.2048.1 protein. The RDC-based structure of PF.2048.1 exhibited 1.0 Å BB-RMSD with respect to a high-quality NOE-based structure. Although optimal strategies would include using RDC data together with chemical shift, NOE, and other NMR data, these studies provide proof-of-principle for robust structure determination of largely-perdeuterated proteins from RDC data alone using REDCRAFT.
核磁共振(NMR)光谱学是表征大分子结构(包括蛋白质结构)的三种主要实验手段之一。传统上,通过溶液NMR光谱学确定结构在很大程度上依赖于源自核Overhauser效应(NOE)测量的距离约束。虽然基于NOE的约束来确定蛋白质结构已得到充分理解并被广泛应用,但基于剩余偶极耦合(RDC)的结构确定相对来说发展得不够完善。在此,我们描述了蛋白质结构建模程序REDCRAFT的新特性,并重点关注新的自适应抽取(AD)特性。AD在提高REDCRAFT对缺失或有噪声数据的鲁棒性方面起着关键作用,同时允许从较少数据确定更大蛋白质的结构。在本报告中,我们展示了REDCRAFT在使用实验收集的数据确定大小从50到145个残基的蛋白质结构,以及使用模拟RDC数据确定更大蛋白质(145到573个残基)结构方面的成功应用。在这两种情况下,REDCRAFT仅使用可从全氘代蛋白质收集的RDC数据。最后,我们将仅基于RDC确定结构的准确性与用于结构新颖的PF.2048.1蛋白质的传统基于NOE的方法进行了比较。PF.2048.1基于RDC的结构相对于高质量的基于NOE的结构表现出1.0 Å的主链均方根偏差(BB-RMSD)。尽管最佳策略将包括结合使用RDC数据与化学位移、NOE和其他NMR数据,但这些研究为仅使用REDCRAFT从RDC数据稳健确定全氘代蛋白质的结构提供了原理证明。