Muñoz-Benavent Maria, Hartkopf Felix, Van Den Bossche Tim, Piro Vitor C, García-Ferris Carlos, Latorre Amparo, Renard Bernhard Y, Muth Thilo
Institute for Integrative Systems Biology (I2SysBio), Universitat de València/CSIC, Paterna (València) 46980, Spain.
Bioinformatics Unit (MF 1), Department for Methods Development and Research Infrastructure, Robert Koch Institute, Berlin 13353, Germany.
NAR Genom Bioinform. 2020 Aug 5;2(3):lqaa058. doi: 10.1093/nargab/lqaa058. eCollection 2020 Sep.
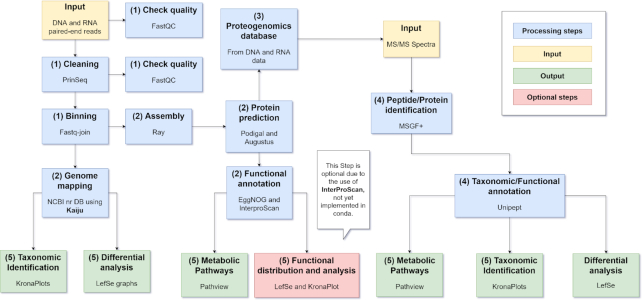
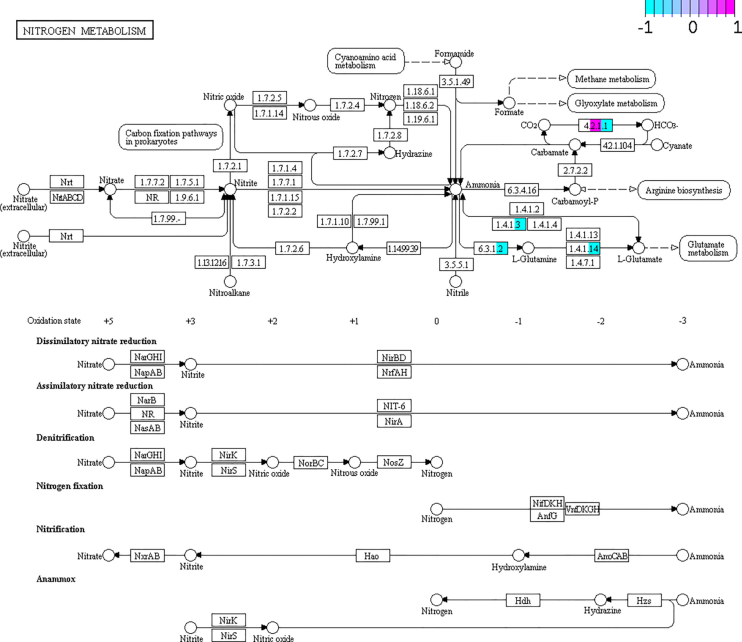
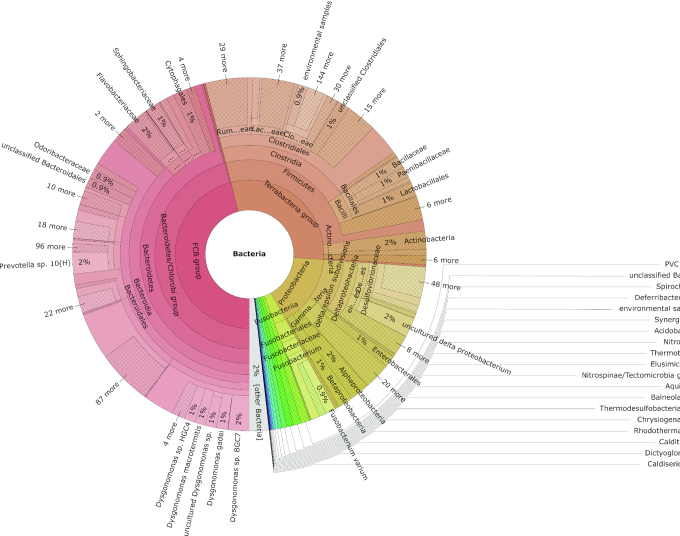
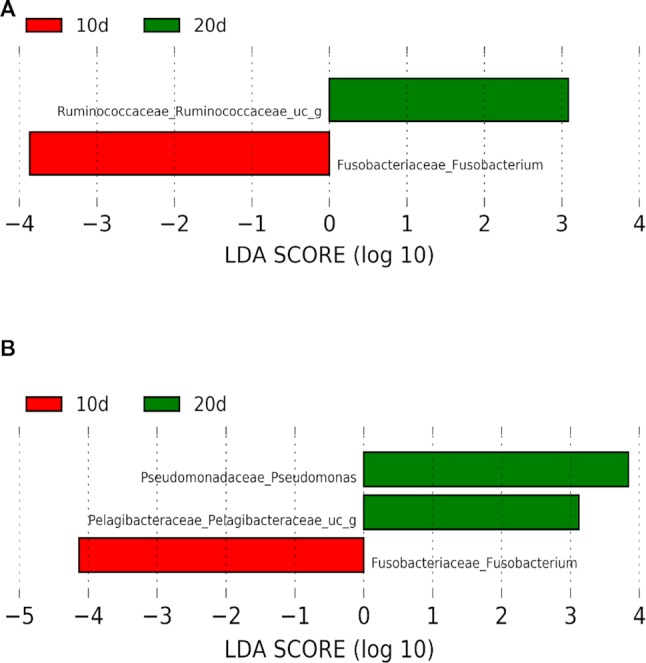
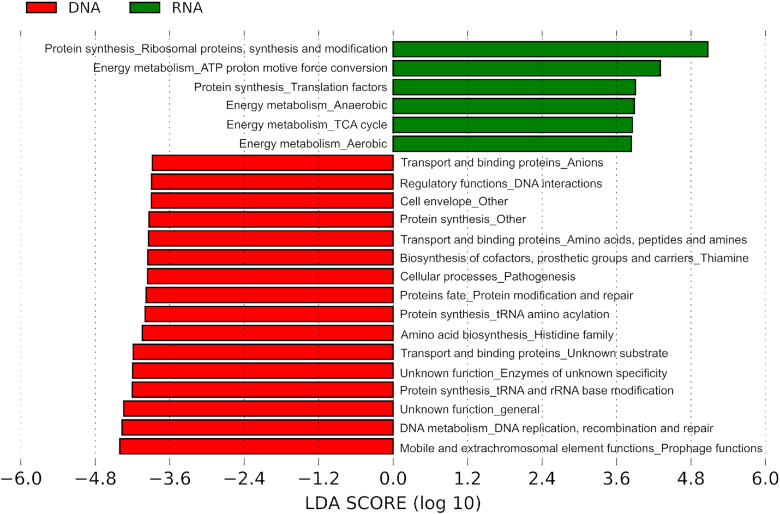
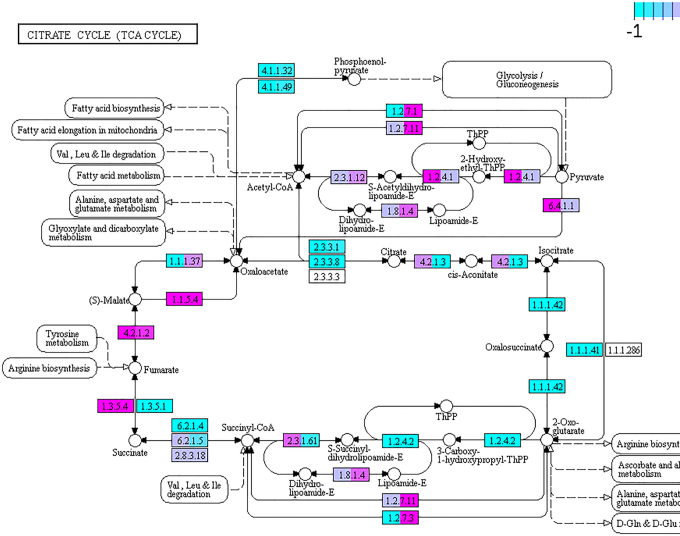
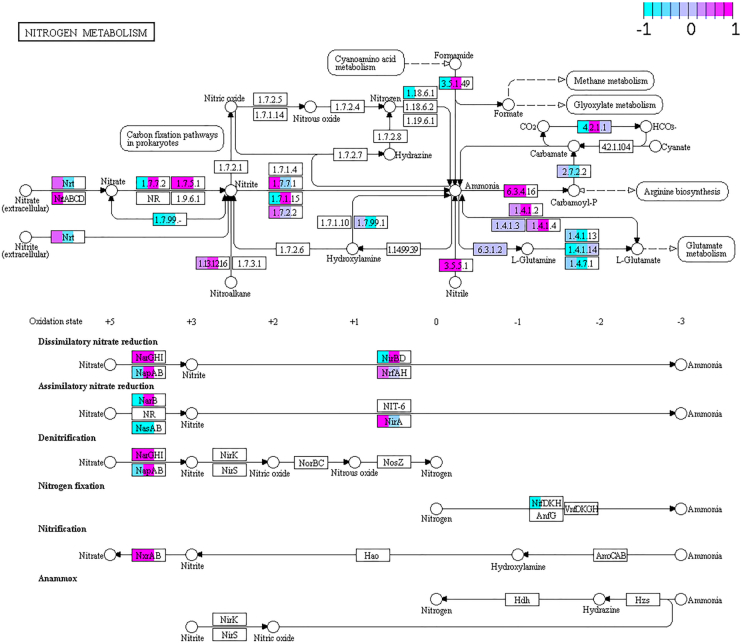
The study of bacterial symbioses has grown exponentially in the recent past. However, existing bioinformatic workflows of microbiome data analysis do commonly not integrate multiple meta-omics levels and are mainly geared toward human microbiomes. Microbiota are better understood when analyzed in their biological context; that is together with their host or environment. Nevertheless, this is a limitation when studying non-model organisms mainly due to the lack of well-annotated sequence references. Here, we present gNOMO, a bioinformatic pipeline that is specifically designed to process and analyze non-model organism samples of up to three meta-omics levels: metagenomics, metatranscriptomics and metaproteomics in an integrative manner. The pipeline has been developed using the workflow management framework Snakemake in order to obtain an automated and reproducible pipeline. Using experimental datasets of the German cockroach , a non-model organism with very complex gut microbiome, we show the capabilities of gNOMO with regard to meta-omics data integration, expression ratio comparison, taxonomic and functional analysis as well as intuitive output visualization. In conclusion, gNOMO is a bioinformatic pipeline that can easily be configured, for integrating and analyzing multiple meta-omics data types and for producing output visualizations, specifically designed for integrating paired-end sequencing data with mass spectrometry from non-model organisms.
近年来,对细菌共生关系的研究呈指数级增长。然而,现有的微生物组数据分析生物信息学工作流程通常并未整合多个元组学水平,且主要针对人类微生物组。在生物学背景下,即与宿主或环境一起分析时,微生物群能得到更好的理解。然而,在研究非模式生物时,这是一个限制,主要原因是缺乏注释良好的序列参考。在这里,我们展示了gNOMO,这是一种生物信息学管道,专门设计用于以综合方式处理和分析多达三个元组学水平的非模式生物样本:宏基因组学、宏转录组学和宏蛋白质组学。该管道是使用工作流管理框架Snakemake开发的,以获得一个自动化且可重复的管道。使用德国小蠊(一种具有非常复杂肠道微生物群的非模式生物)的实验数据集,我们展示了gNOMO在元组学数据整合、表达比率比较分类学和功能分析以及直观输出可视化方面的能力。总之,gNOMO是一种生物信息学管道,可轻松配置,用于整合和分析多种元组学数据类型并生成输出可视化,专门设计用于整合来自非模式生物的双端测序数据和质谱数据。