Chen Shengquan, Gan Mingxin, Lv Hairong, Jiang Rui
Ministry of Education Key Laboratory of Bioinformatics, Bioinformatics Division at the Beijing National Research Center for Information Science and Technology, Center for Synthetic and Systems Biology, Department of Automation, Tsinghua University, Beijing 100084, China.
Department of Management Science and Engineering, School of Economics and Management, University of Science and Technology Beijing, Beijing 100083, China.
Genomics Proteomics Bioinformatics. 2021 Aug;19(4):565-577. doi: 10.1016/j.gpb.2019.04.006. Epub 2021 Feb 11.
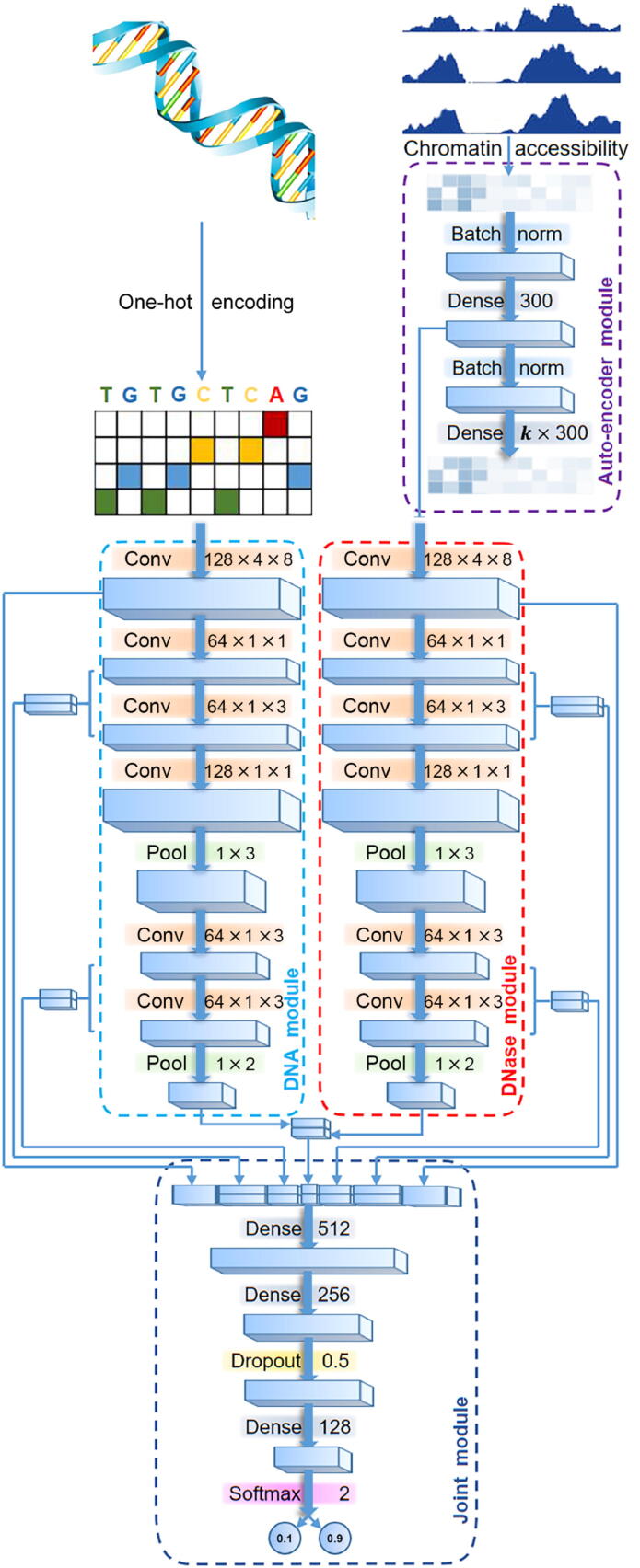
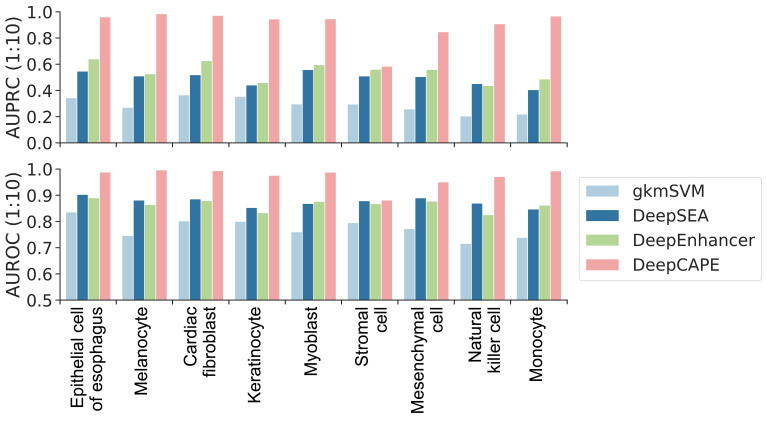
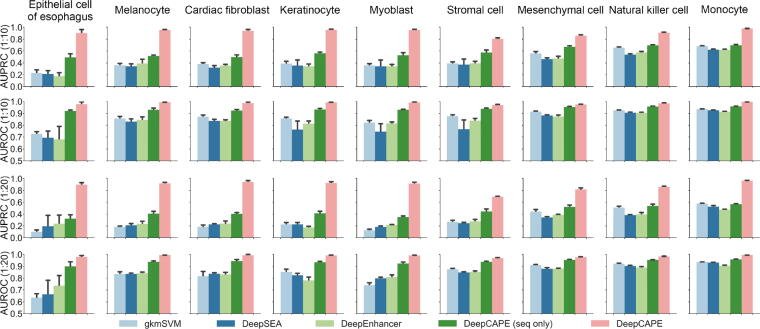
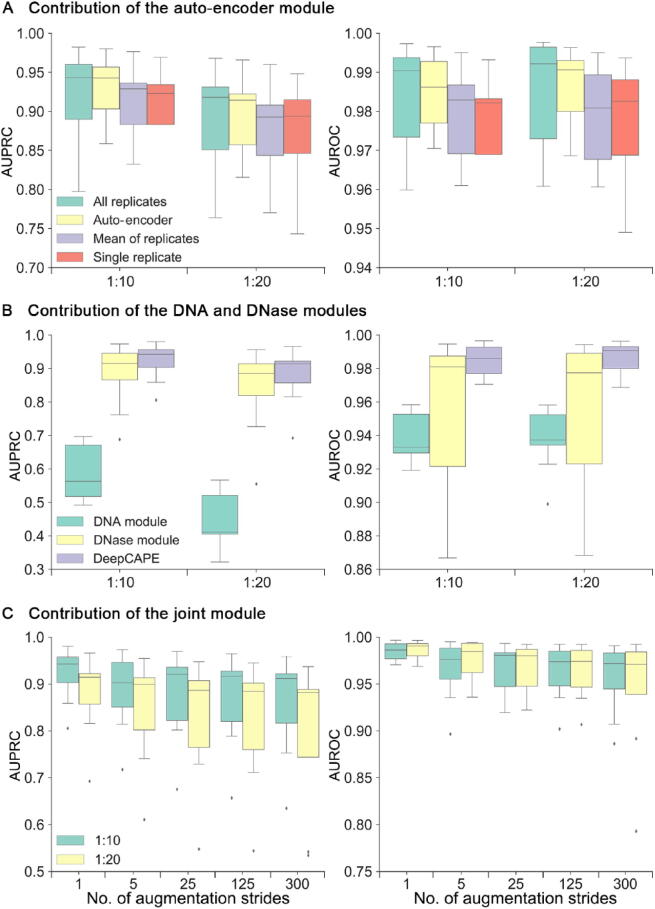
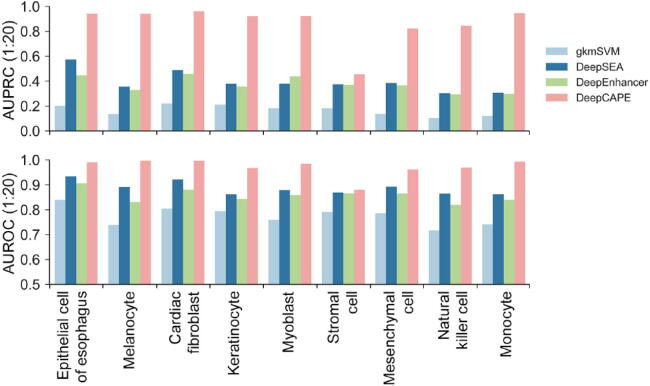
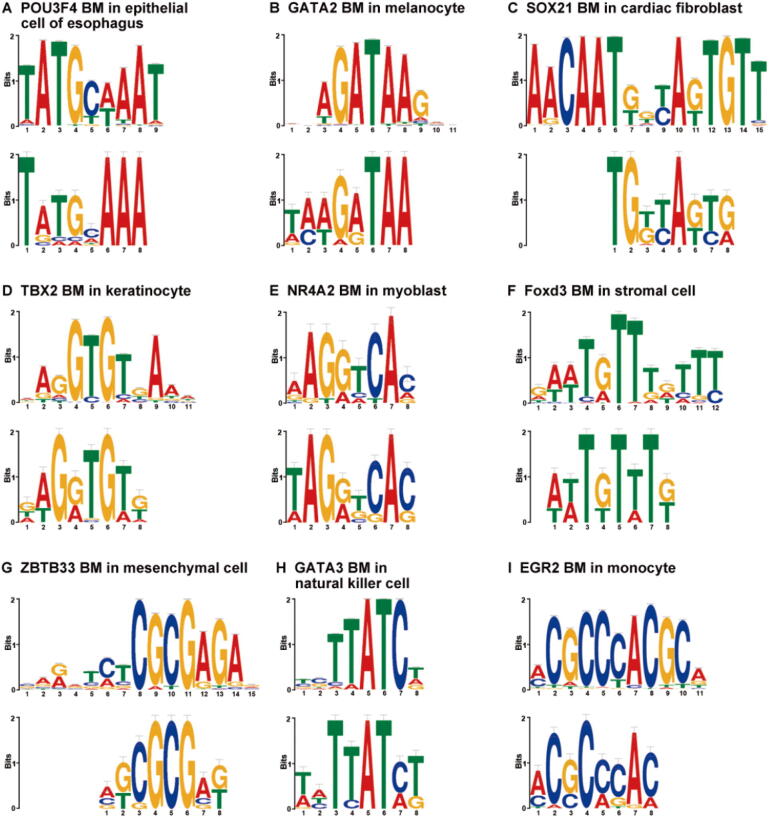
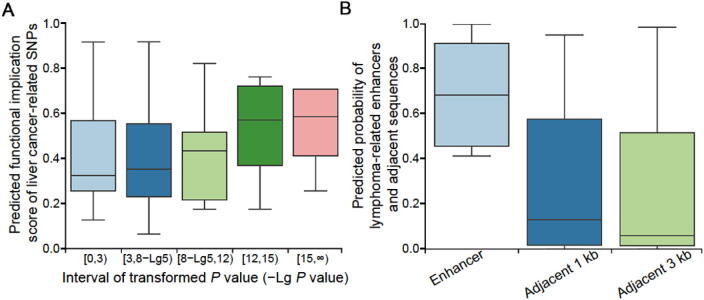
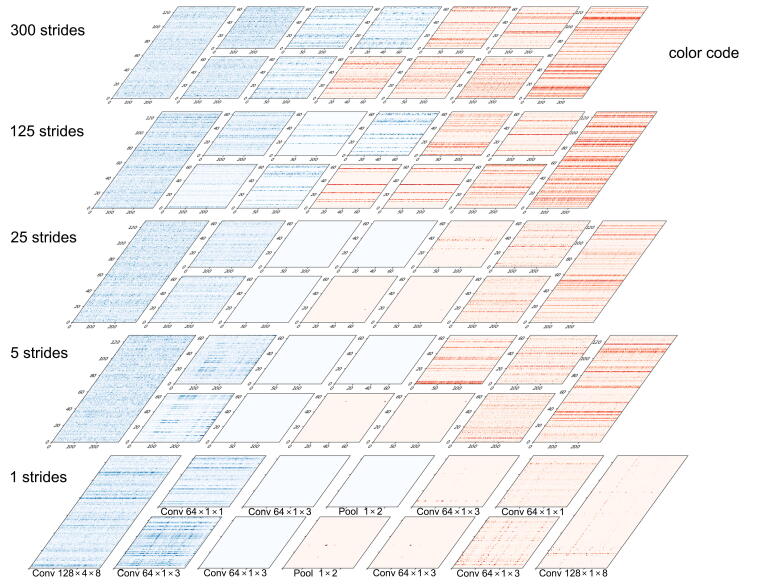
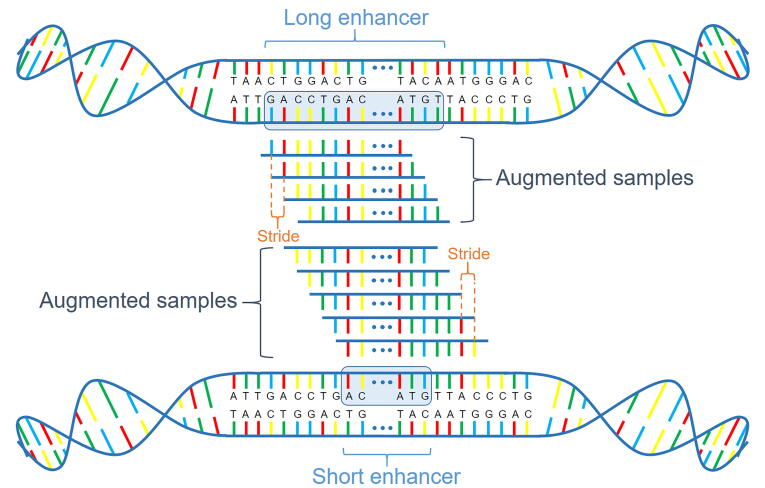
The establishment of a landscape of enhancers across human cells is crucial to deciphering the mechanism of gene regulation, cell differentiation, and disease development. High-throughput experimental approaches, which contain successfully reported enhancers in typical cell lines, are still too costly and time-consuming to perform systematic identification of enhancers specific to different cell lines. Existing computational methods, capable of predicting regulatory elements purely relying on DNA sequences, lack the power of cell line-specific screening. Recent studies have suggested that chromatin accessibility of a DNA segment is closely related to its potential function in regulation, and thus may provide useful information in identifying regulatory elements. Motivated by the aforementioned understanding, we integrate DNA sequences and chromatin accessibility data to accurately predict enhancers in a cell line-specific manner. We proposed DeepCAPE, a deep convolutional neural network to predict enhancers via the integration of DNA sequences and DNase-seq data. Benefitting from the well-designed feature extraction mechanism and skip connection strategy, our model not only consistently outperforms existing methods in the imbalanced classification of cell line-specific enhancers against background sequences, but also has the ability to self-adapt to different sizes of datasets. Besides, with the adoption of auto-encoder, our model is capable of making cross-cell line predictions. We further visualize kernels of the first convolutional layer and show the match of identified sequence signatures and known motifs. We finally demonstrate the potential ability of our model to explain functional implications of putative disease-associated genetic variants and discriminate disease-related enhancers. The source code and detailed tutorial of DeepCAPE are freely available at https://github.com/ShengquanChen/DeepCAPE.
绘制人类细胞中增强子图谱对于解读基因调控、细胞分化和疾病发展机制至关重要。高通量实验方法虽已成功报道了典型细胞系中的增强子,但进行不同细胞系特异性增强子的系统鉴定仍成本高昂且耗时。现有的计算方法仅依靠DNA序列预测调控元件,缺乏细胞系特异性筛选能力。近期研究表明,DNA片段的染色质可及性与其潜在调控功能密切相关,可为鉴定调控元件提供有用信息。基于上述认识,我们整合DNA序列和染色质可及性数据,以细胞系特异性方式准确预测增强子。我们提出了DeepCAPE,一种通过整合DNA序列和DNase-seq数据来预测增强子的深度卷积神经网络。受益于精心设计的特征提取机制和跳跃连接策略,我们的模型不仅在细胞系特异性增强子与背景序列的不平衡分类中始终优于现有方法,还能够自适应不同规模的数据集。此外,通过采用自动编码器,我们的模型能够进行跨细胞系预测。我们进一步可视化了第一层卷积层的内核,并展示了识别出的序列特征与已知基序的匹配情况。我们最终证明了我们的模型在解释假定的疾病相关遗传变异功能影响和区分疾病相关增强子方面的潜在能力。DeepCAPE的源代码和详细教程可在https://github.com/ShengquanChen/DeepCAPE上免费获取。