Ministry of Education Key Laboratory of Bioinformatics, Bioinformatics Division at the Beijing National Research Center for Information Science and Technology, Center for Synthetic and Systems Biology, Department of Automation, Tsinghua University, Beijing, China.
Department of Statistics, The Chinese University of Hong Kong, Hong Kong SAR, China.
Nat Commun. 2021 Apr 12;12(1):2177. doi: 10.1038/s41467-021-22495-4.
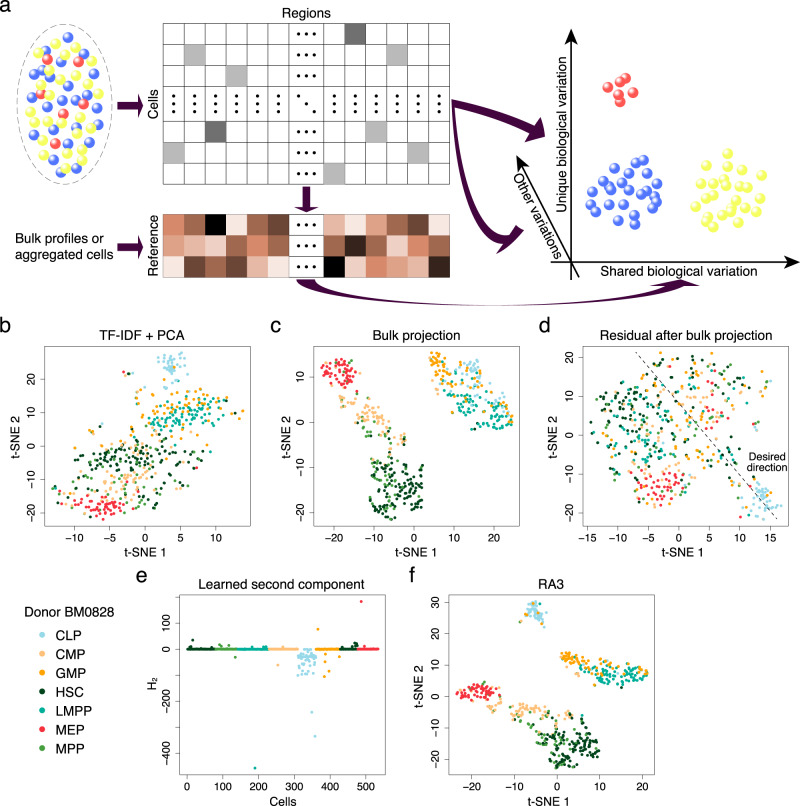
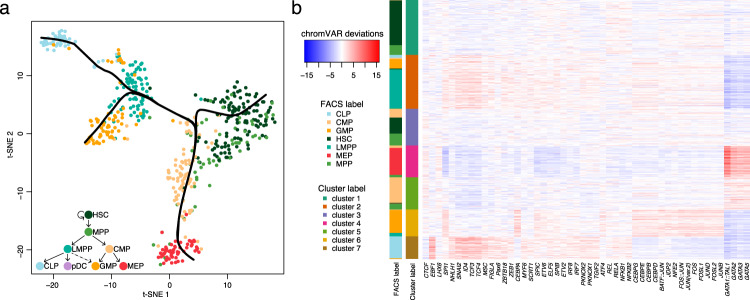
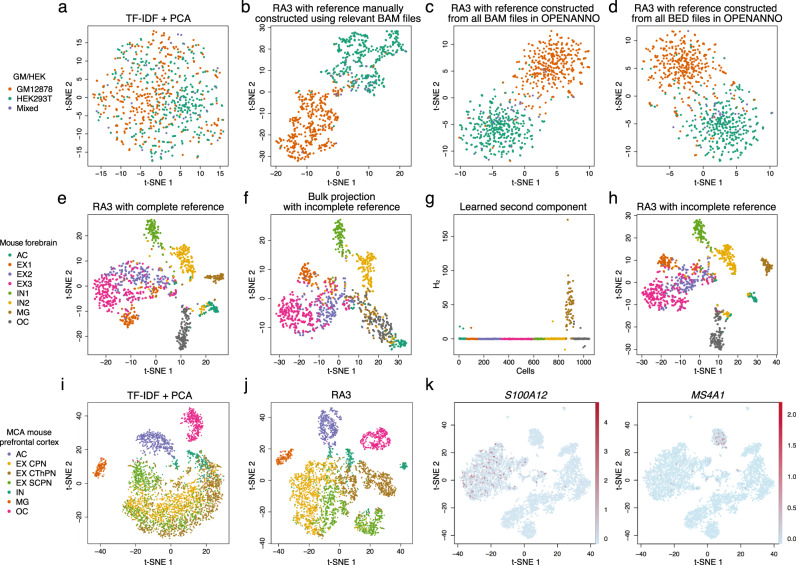
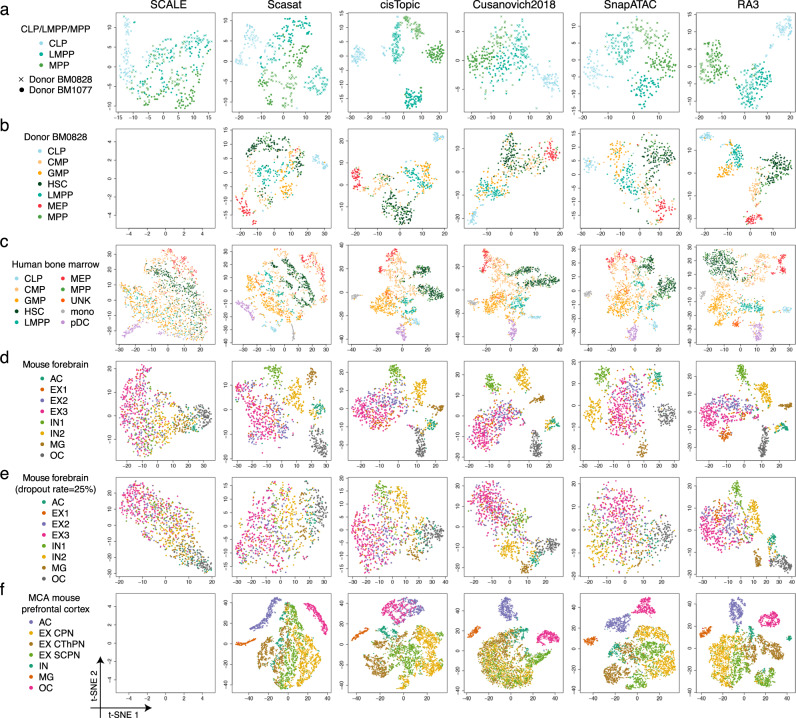
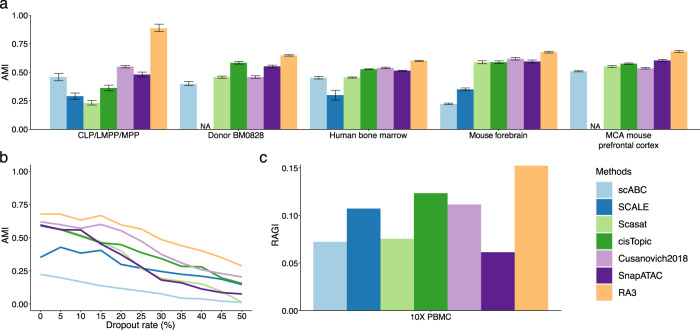
The recent advancements in single-cell technologies, including single-cell chromatin accessibility sequencing (scCAS), have enabled profiling the epigenetic landscapes for thousands of individual cells. However, the characteristics of scCAS data, including high dimensionality, high degree of sparsity and high technical variation, make the computational analysis challenging. Reference-guided approaches, which utilize the information in existing datasets, may facilitate the analysis of scCAS data. Here, we present RA3 (Reference-guided Approach for the Analysis of single-cell chromatin Accessibility data), which utilizes the information in massive existing bulk chromatin accessibility and annotated scCAS data. RA3 simultaneously models (1) the shared biological variation among scCAS data and the reference data, and (2) the unique biological variation in scCAS data that identifies distinct subpopulations. We show that RA3 achieves superior performance when used on several scCAS datasets, and on references constructed using various approaches. Altogether, these analyses demonstrate the wide applicability of RA3 in analyzing scCAS data.
单细胞技术的最新进展,包括单细胞染色质可及性测序(scCAS),使对数千个单个细胞的表观基因组景观进行分析成为可能。然而,scCAS 数据的特征,包括高维度、高度稀疏性和高度技术变化,使得计算分析具有挑战性。参考引导方法利用现有数据集的信息,可以促进 scCAS 数据的分析。在这里,我们提出了 RA3(用于分析单细胞染色质可及性数据的参考引导方法),它利用了大量现有的批量染色质可及性和注释 scCAS 数据中的信息。RA3 同时对以下内容进行建模:(1) scCAS 数据和参考数据之间的共享生物学变异,以及 (2) scCAS 数据中的独特生物学变异,这些变异可以识别不同的亚群。我们表明,RA3 在几个 scCAS 数据集和使用各种方法构建的参考上的性能表现优异。总之,这些分析表明 RA3 在分析 scCAS 数据方面具有广泛的适用性。