Department of Computer and Electrical Engineering and Computer Science, Florida Atlantic University, Boca Raton, FL 33431, USA.
Department of Biology, Texas A&M University, College Station, TX 77843, USA.
Syst Biol. 2021 Jun 16;70(4):660-680. doi: 10.1093/sysbio/syab009.
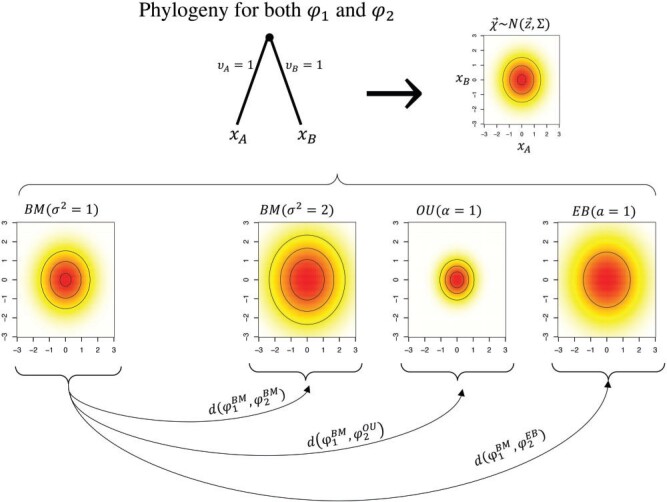
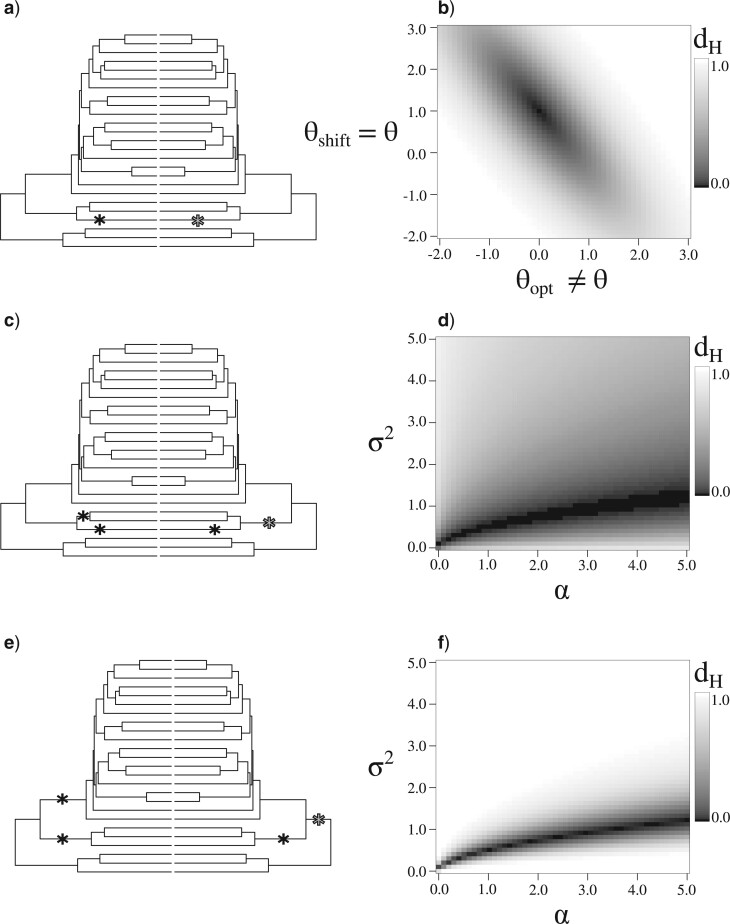
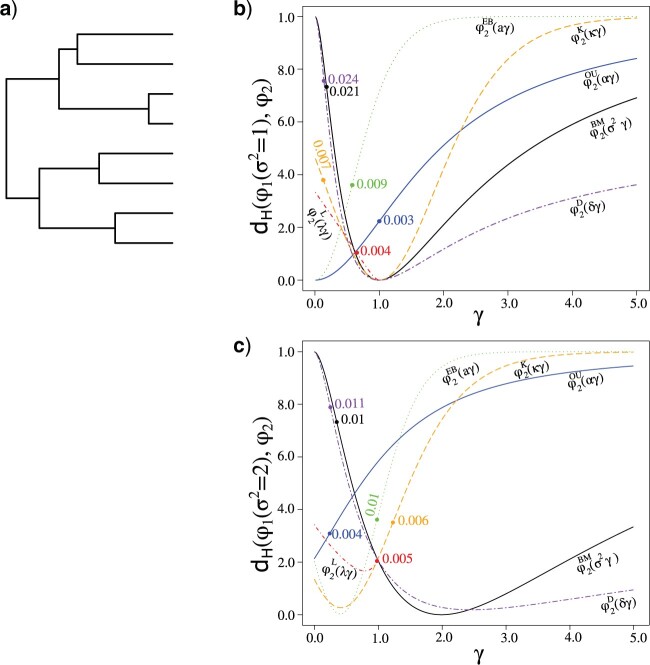
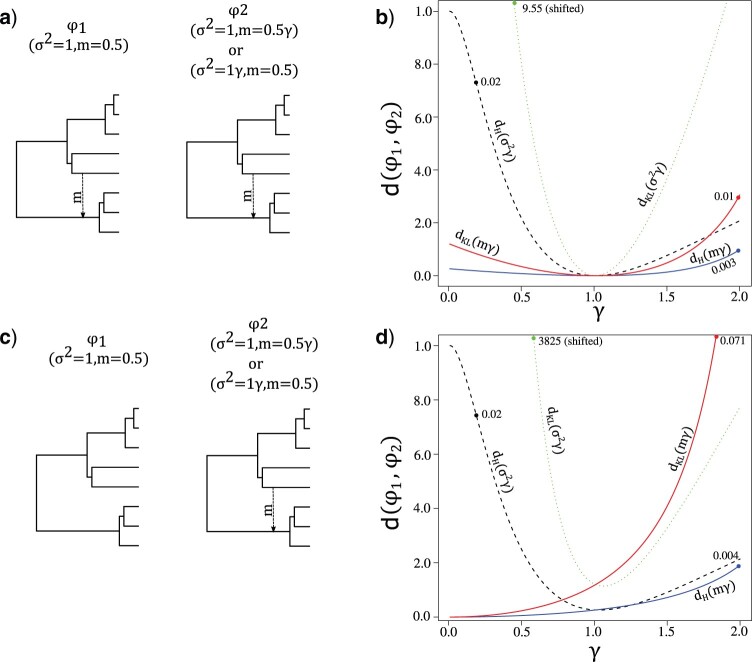
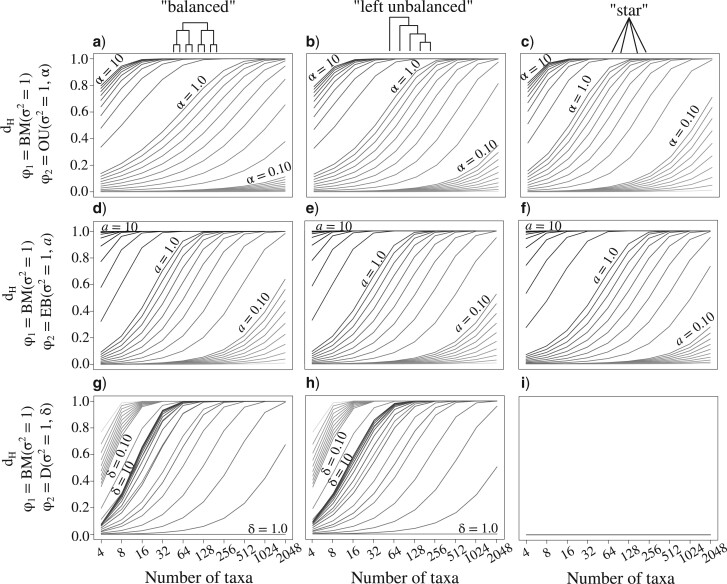
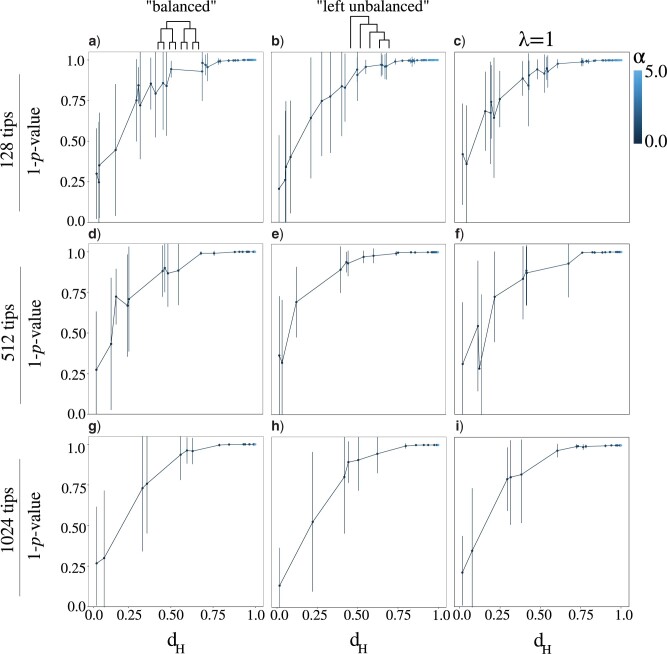
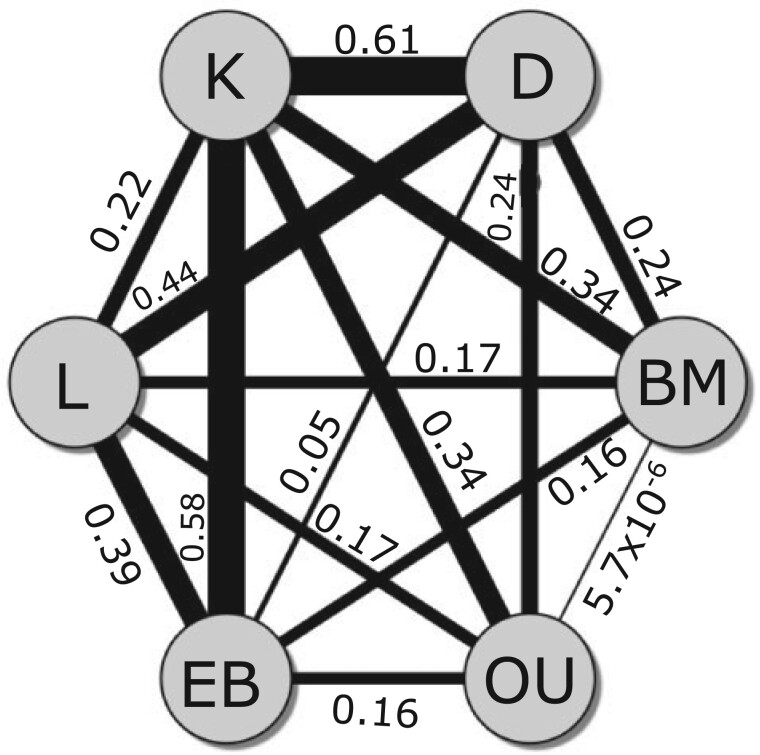
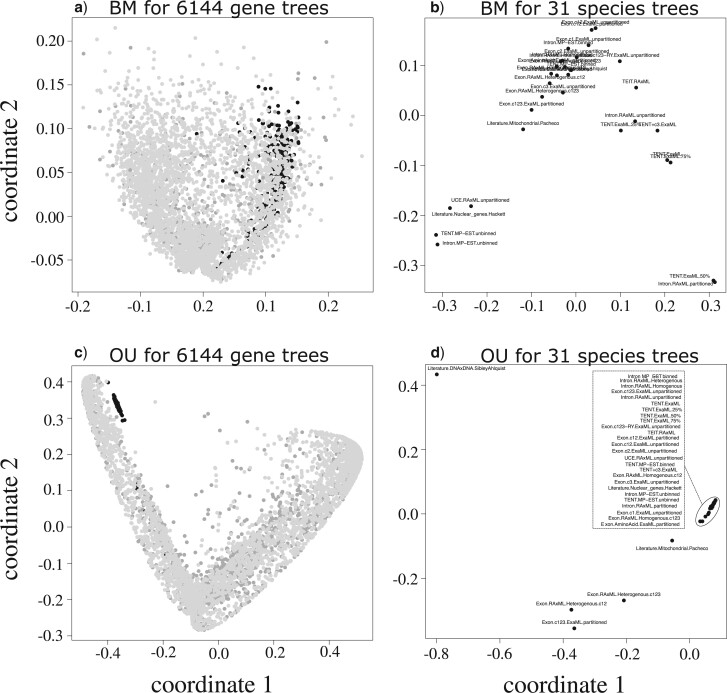
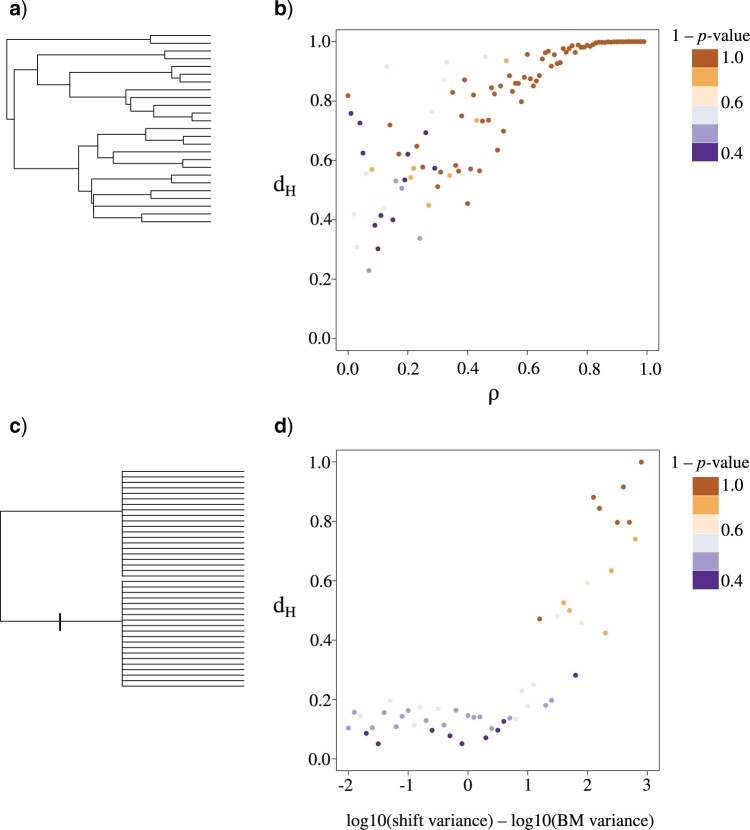
Stochastic models of character trait evolution have become a cornerstone of evolutionary biology in an array of contexts. While probabilistic models have been used extensively for statistical inference, they have largely been ignored for the purpose of measuring distances between phylogeny-aware models. Recent contributions to the problem of phylogenetic distance computation have highlighted the importance of explicitly considering evolutionary model parameters and their impacts on molecular sequence data when quantifying dissimilarity between trees. By comparing two phylogenies in terms of their induced probability distributions that are functions of many model parameters, these distances can be more informative than traditional approaches that rely strictly on differences in topology or branch lengths alone. Currently, however, these approaches are designed for comparing models of nucleotide substitution and gene tree distributions, and thus, are unable to address other classes of traits and associated models that may be of interest to evolutionary biologists. Here, we expand the principles of probabilistic phylogenetic distances to compute tree distances under models of continuous trait evolution along a phylogeny. By explicitly considering both the degree of relatedness among species and the evolutionary processes that collectively give rise to character traits, these distances provide a foundation for comparing models and their predictions, and for quantifying the impacts of assuming one phylogenetic background over another while studying the evolution of a particular trait. We demonstrate the properties of these approaches using theory, simulations, and several empirical data sets that highlight potential uses of probabilistic distances in many scenarios. We also introduce an open-source R package named PRDATR for easy application by the scientific community for computing phylogenetic distances under models of character trait evolution.[Brownian motion; comparative methods; phylogeny; quantitative traits.].
随机特质进化模型已经成为诸多情境下进化生物学的基石。概率模型虽然被广泛用于统计推断,但在测量与系统发育相关模型之间的距离方面,它们在很大程度上被忽视了。最近,在计算系统发生距离的问题上的一些进展强调了在量化树之间的差异时,明确考虑进化模型参数及其对分子序列数据的影响的重要性。通过比较两个系统发生树的诱导概率分布,这些概率分布是许多模型参数的函数,这些距离比仅依赖拓扑或分支长度差异的传统方法更具信息量。然而,目前这些方法是为比较核苷酸替代和基因树分布模型而设计的,因此无法解决进化生物学家可能感兴趣的其他类别的特质和相关模型。在这里,我们将概率系统发生距离的原理扩展到计算系统发生树上连续特质进化模型下的树距离。通过明确考虑物种之间的亲缘关系程度以及共同导致特质的进化过程,这些距离为比较模型及其预测提供了基础,并量化了在研究特定特质的进化时,假设另一个系统发生背景相对于另一个系统发生背景的影响。我们使用理论、模拟和几个实证数据集来证明这些方法的特性,这些数据集突出了在许多情况下概率距离在许多场景中的潜在用途。我们还引入了一个名为 PRDATR 的开源 R 包,方便科学界在特质进化模型下计算系统发生距离。[布朗运动;比较方法;系统发生;数量特质。]。