Department of Ecology, University of Innsbruck, Technikerstraße 25, 6020, Innsbruck, Austria.
Department of Botany, University of Innsbruck, Sternwartestraße 15, 6020, Innsbruck, Austria.
Sci Rep. 2021 Feb 17;11(1):3978. doi: 10.1038/s41598-020-79778-x.
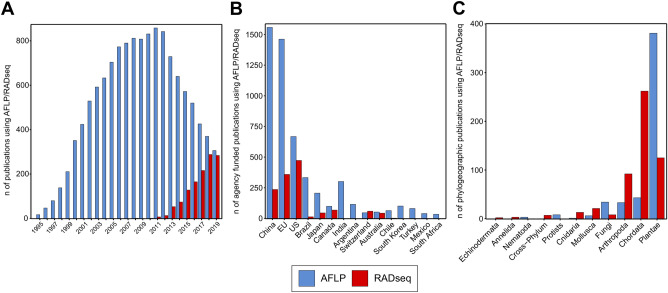
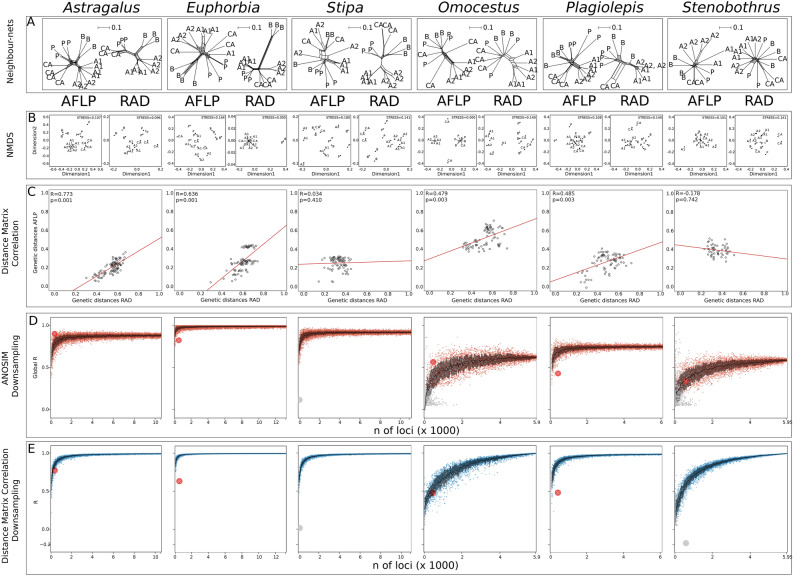
Multi-locus genetic data are pivotal in phylogenetics. Today, high-throughput sequencing (HTS) allows scientists to generate an unprecedented amount of such data from any organism. However, HTS is resource intense and may not be accessible to wide parts of the scientific community. In phylogeography, the use of HTS has concentrated on a few taxonomic groups, and the amount of data used to resolve a phylogeographic pattern often seems arbitrary. We explore the performance of two genetic marker sampling strategies and the effect of marker quantity in a comparative phylogeographic framework focusing on six species (arthropods and plants). The same analyses were applied to data inferred from amplified fragment length polymorphism fingerprinting (AFLP), a cheap, non-HTS based technique that is able to straightforwardly produce several hundred markers, and from restriction site associated DNA sequencing (RADseq), a more expensive, HTS-based technique that produces thousands of single nucleotide polymorphisms. We show that in four of six study species, AFLP leads to results comparable with those of RADseq. While we do not aim to contest the advantages of HTS techniques, we also show that AFLP is a robust technique to delimit evolutionary entities in both plants and animals. The demonstrated similarity of results from the two techniques also strengthens biological conclusions that were based on AFLP data in the past, an important finding given the wide utilization of AFLP over the last decades. We emphasize that whenever the delimitation of evolutionary entities is the central goal, as it is in many fields of biodiversity research, AFLP is still an adequate technique.
多基因座遗传数据在系统发育学中至关重要。如今,高通量测序(HTS)使科学家能够从任何生物体中生成前所未有的此类数据。然而,HTS 资源密集,可能无法为科学界的广泛群体所接受。在系统地理学中,HTS 的使用集中在少数几个分类群上,用于解决系统地理学模式的数据量似乎往往是任意的。我们在一个比较系统地理学框架中探讨了两种遗传标记采样策略的性能和标记数量的影响,该框架侧重于六个物种(节肢动物和植物)。相同的分析应用于从扩增片段长度多态性指纹图谱(AFLP)推断的数据,这是一种廉价的非 HTS 技术,能够直接产生数百个标记,以及从限制性位点相关 DNA 测序(RADseq)推断的数据,这是一种更昂贵的 HTS 技术,可产生数千个单核苷酸多态性。我们表明,在六个研究物种中的四个物种中,AFLP 导致的结果与 RADseq 的结果相当。虽然我们并不旨在质疑 HTS 技术的优势,但我们也表明 AFLP 是一种在植物和动物中划分进化实体的稳健技术。这两种技术的结果相似也加强了过去基于 AFLP 数据得出的生物学结论,这是一个重要的发现,因为在过去几十年中 AFLP 被广泛应用。我们强调,只要进化实体的划界是中心目标,就像在生物多样性研究的许多领域一样,AFLP 仍然是一种合适的技术。