Key Laboratory of Forest Genetics & Biotechnology of Ministry of Education, Co-Innovation Center for Sustainable Forestry in Southern China, Nanjing Forestry University, Nanjing 210037, China.
School of Animal Science and Technology, Jingling Institute of Technology, Nanjing 210038, China.
G3 (Bethesda). 2021 Feb 9;11(2). doi: 10.1093/g3journal/jkaa053.
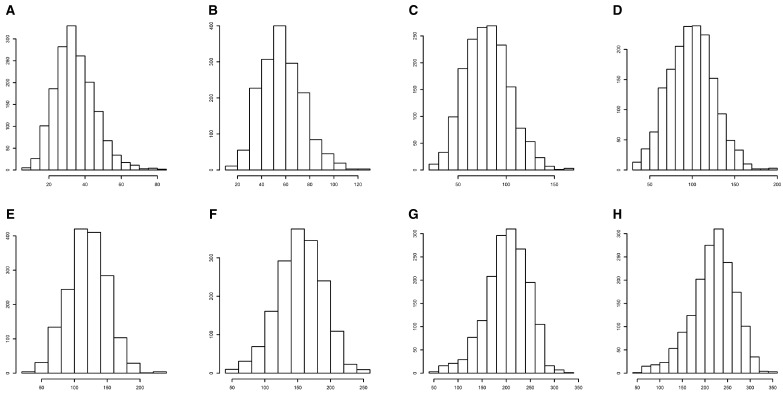
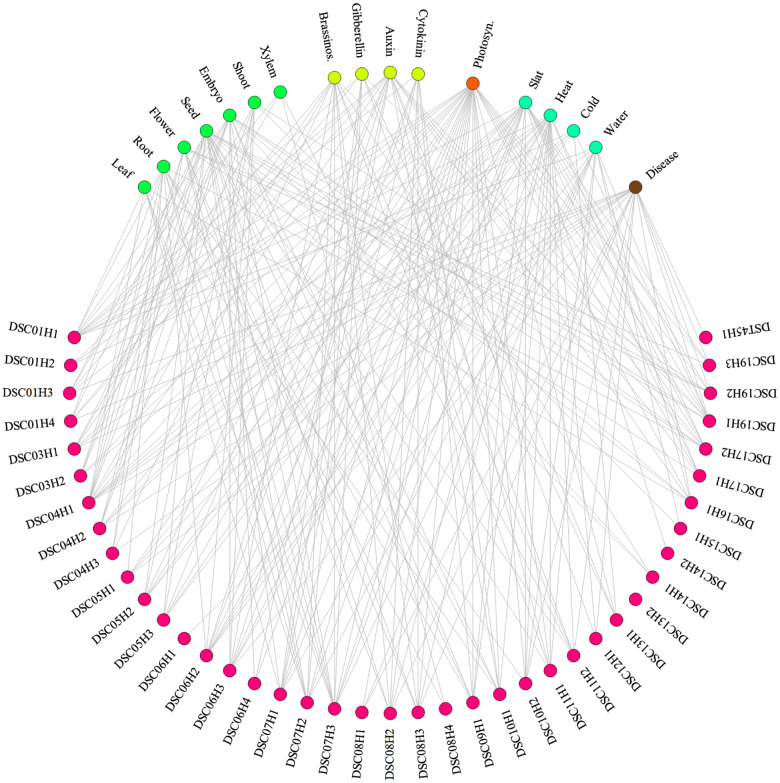
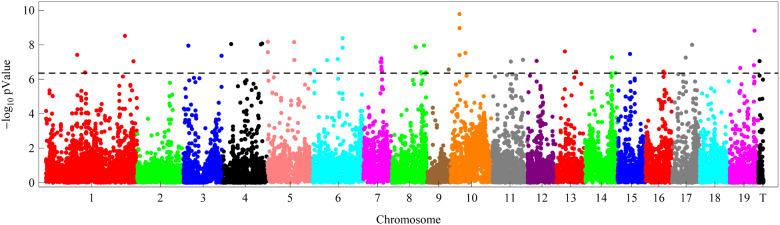
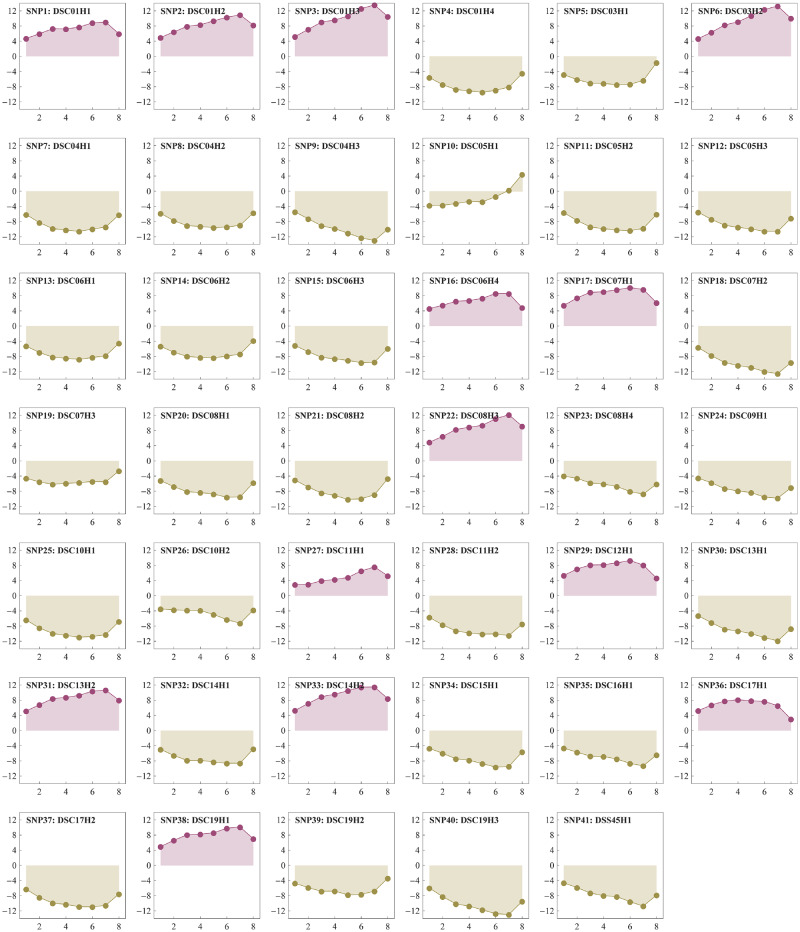
With the advances in high-throughput sequencing technologies, it is not difficult to extract tens of thousands of single-nucleotide polymorphisms (SNPs) across many individuals in a fast and cheap way, making it possible to perform genome-wide association studies (GWAS) of quantitative traits in outbred forest trees. It is very valuable to apply traditional breeding experiments in GWAS for identifying genome variants associated with ecologically and economically important traits in Populus. Here, we reported a GWAS of tree height measured at multiple time points from a randomized complete block design (RCBD), which was established with clones from an F1 hybrid population of Populus deltoides and Populus simonii. A total of 22,670 SNPs across 172 clones in the RCBD were obtained with restriction site-associated DNA sequencing (RADseq) technology. The multivariate mixed linear model was applied by incorporating the pedigree relationship matrix of individuals to test the association of each SNP to the tree heights over 8 time points. Consequently, 41 SNPs were identified significantly associated with the tree height under the P-value threshold determined by Bonferroni correction at the significant level of 0.01. These SNPs were distributed on all but two chromosomes (Chr02 and Chr18) and explained the phenotypic variance ranged from 0.26% to 2.64%, amounting to 63.68% in total. Comparison with previous mapping studies for poplar height as well as the candidate genes of these detected SNPs were also investigated. We therefore showed that the application of multivariate linear mixed model to the longitudinal phenotypic data from the traditional breeding experimental design facilitated to identify far more genome-wide variants for tree height in poplar. The significant SNPs identified in this study would enhance understanding of molecular mechanism for growth traits and would accelerate marker-assisted breeding programs in Populus.
利用高通量测序技术的进步,以快速且廉价的方式从许多个体中提取数万个性单核苷酸多态性(SNP)并不困难,这使得在外生树木中进行全基因组关联研究(GWAS)变得可行。在外生树木中应用传统的 GWAS 来鉴定与生态和经济重要性状相关的基因组变异是非常有价值的。在这里,我们报告了一项针对柳树和杨树杂交 F1 群体的无性系随机完全区组设计(RCBD)中多个时间点测量的树高的 GWAS。利用 RADseq 技术共获得了 RCBD 中 172 个无性系中的 22670 个 SNP。通过纳入个体的系谱关系矩阵,应用多元混合线性模型来检验每个 SNP 与 8 个时间点的树高之间的关联。结果,在通过 Bonferroni 校正确定的 P 值阈值下,有 41 个 SNP 与树高显著相关,达到了 0.01 的显著水平。这些 SNP 分布在除 Chr02 和 Chr18 之外的所有染色体上,解释了表型方差的范围从 0.26%到 2.64%,总计为 63.68%。还对柳树高度的先前作图研究以及检测到的这些 SNP 的候选基因进行了比较。因此,我们表明,将多元线性混合模型应用于传统育种实验设计的纵向表型数据,有利于鉴定柳树全基因组范围内更多与树高相关的变异。本研究中鉴定的显著 SNP 将有助于深入了解生长性状的分子机制,并加速杨树的标记辅助育种计划。