Istituto per la Sintesi Organica e la Fotoreattività, Consiglio Nazionale delle Ricerche (ISOF-CNR), Via P. Gobetti 101, 40129 Bologna, Italy.
Dipartimento di Chimica Industriale "Toso Montanari", Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy.
Acc Chem Res. 2021 Mar 16;54(6):1492-1505. doi: 10.1021/acs.accounts.0c00825. Epub 2021 Feb 22.
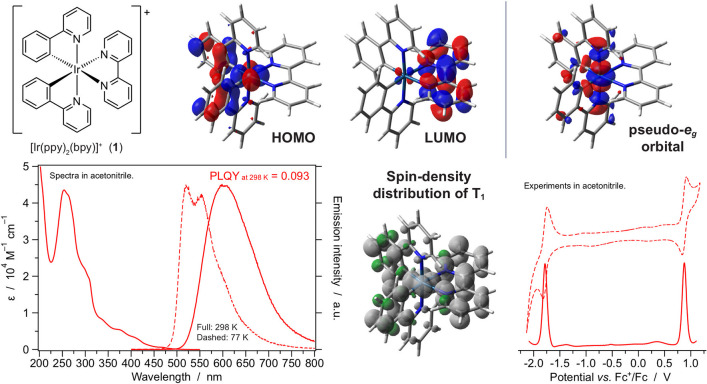
Iridium(III) complexes have assumed a prominent role in the areas of photochemistry and photophysics due to the peculiar properties of both the metal itself and the ligand environment that can be assembled around it. Ir(III) is larger, heavier, and bears a higher ionic charge than its analogue and widely used d ions such as Fe(II) and Ru(II). Accordingly, its complexes exhibit wider ligand-field d-d orbital splitting with electronic levels centered on the metal, typically nonemissive and photodissociative, not playing a relevant role in excited-state deactivations. In other words, iridium complexes are typically more stable and/or more emissive than Fe(II) and Ru(II) systems. Additionally, the particularly strong heavy-atom effect of iridium promotes singlet-triplet transitions, with characteristic absorption features in the UV-vis and relatively short excited-state lifetimes of emissive triplet levels. Ir(III) is also a platform for anchoring ligands of rather different sorts. Its versatile chemistry includes not only coordination with classic NN neutral ligands but also the binding of negatively charged chelators, typically having a cyclometalating CN anchor. The carbon-metal bond in these systems has some degree of covalent character, but this does not preclude a localized description of the excited states of the related complexes, which can be designated as metal-centered (MC), ligand-centered (LC), or charge transfer (CT), allowing a simplified description of electronic and photophysical properties. The possibility of binding different types of ligands and making heteroleptic complexes is a formidable tool for finely tuning the nature and energy of the lowest electronic excited state of cationic Ir(III) complexes by ligand design. Herein we give an account of our work on several families of iridium complexes typically equipped with two cyclometalating bidentate ligands (CN), in combination with mono or bidentate "ancillary" ligands with NN, CN, and CC motifs. We have explored new synthesis routes for both cyclometalating and ancillary ligands, obtaining primarily cationic complexes but also some neutral or even negatively charged systems. In the domain of the ancillary ligands, we have explored isocyanides, carbenes, mesoionic triazolylidenes, and bis-tetrazolic ligands. For the cyclometalating moiety, we have investigated carbene, mesoionic triazolylidene, and tetrazolic systems. Key results of our work include new strategies to modify both cyclometalating and ancillary ligands by relocating ionic charges, the determination of new factors affecting the stability of complexes, a demonstration of subtle structural effects that strongly modify the photophysical properties, new options to get blue-greenish emitters for optoelectronic devices, and a set of ligand modifications allowing the optimization of electrochemical and excited-state properties to obtain new promising Ir(III) complexes for photoredox catalysis. These results constitute a step forward in the preparation of custom iridium-based materials crafted by excited-state engineering, which is achieved through the concerted effort of computational and synthetic chemistry along with electrochemistry and photochemistry.
三碘化铱(Ir(III))配合物因其金属本身和配位环境的独特性质,在光化学和光物理领域中扮演着重要角色。Ir(III)比其类似物和广泛使用的 d 离子(如 Fe(II)和 Ru(II))更大、更重且带有更高的离子电荷。因此,其配合物表现出更宽的配体场 d-d 轨道分裂,电子能级位于金属中心,通常是非辐射和光解的,在激发态失活过程中不起重要作用。换句话说,铱配合物通常比 Fe(II)和 Ru(II)体系更稳定和/或更具发光性。此外,铱的特殊重原子效应促进了单重态-三重态跃迁,在紫外可见区具有特征吸收特征,并且发射三重态能级的激发态寿命相对较短。Ir(III)也是锚定各种配体的平台。其多功能化学不仅包括与经典 NN 中性配体的配位,还包括与带负电荷的螯合剂的结合,通常具有环金属化的 CN 锚。这些体系中的碳-金属键具有一定程度的共价性质,但这并不排除相关配合物的激发态可以用局域描述,这些激发态可以被指定为金属中心(MC)、配体中心(LC)或电荷转移(CT),从而简化对电子和光物理性质的描述。通过配体设计,可以结合不同类型的配体并形成杂配配合物,这是一种精细调节阳离子 Ir(III)配合物最低电子激发态性质和能量的强大工具。本文介绍了我们在一系列典型的配备两个环金属化双齿配体(CN)的铱配合物方面的工作,同时结合了单齿或双齿“辅助”配体的 NN、CN 和 CC 基序。我们探索了环金属化和辅助配体的新合成途径,主要得到了阳离子配合物,但也得到了一些中性甚至带负电荷的体系。在辅助配体领域,我们探索了异氰化物、卡宾、中氮茚基和双四唑配体。对于环金属化部分,我们研究了卡宾、中氮茚基和四唑体系。我们工作的关键结果包括通过重新定位离子电荷来修饰环金属化和辅助配体的新策略、确定影响配合物稳定性的新因素、证明强烈改变光物理性质的微妙结构效应、获得用于光电设备的蓝绿色发射器的新选择,以及一套允许优化电化学和激发态性质以获得用于光还原催化的新有前途的 Ir(III)配合物的配体修饰。这些结果是通过计算化学和合成化学以及电化学和光化学的协同努力,在制备通过激发态工程定制的基于铱的材料方面迈出的一步,该工程实现了定制的基于铱的材料的制备。