Institute of Hypertension, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China.
Department of Internal Medicine, University of Utah and Veterans Affairs Medical Center, Salt Lake City, Utah, U.S.A.
Clin Sci (Lond). 2021 Mar 26;135(6):793-810. doi: 10.1042/CS20201047.
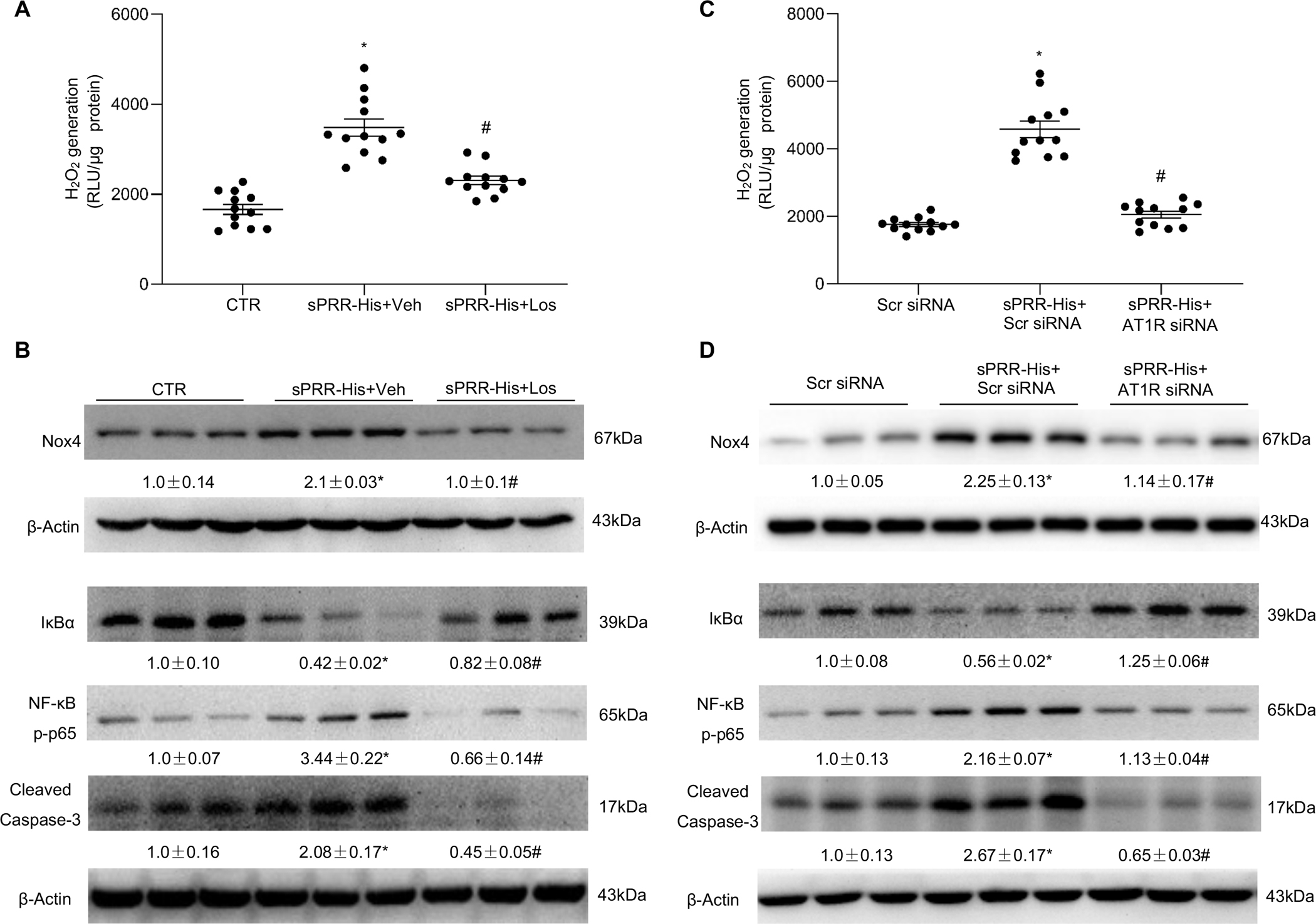
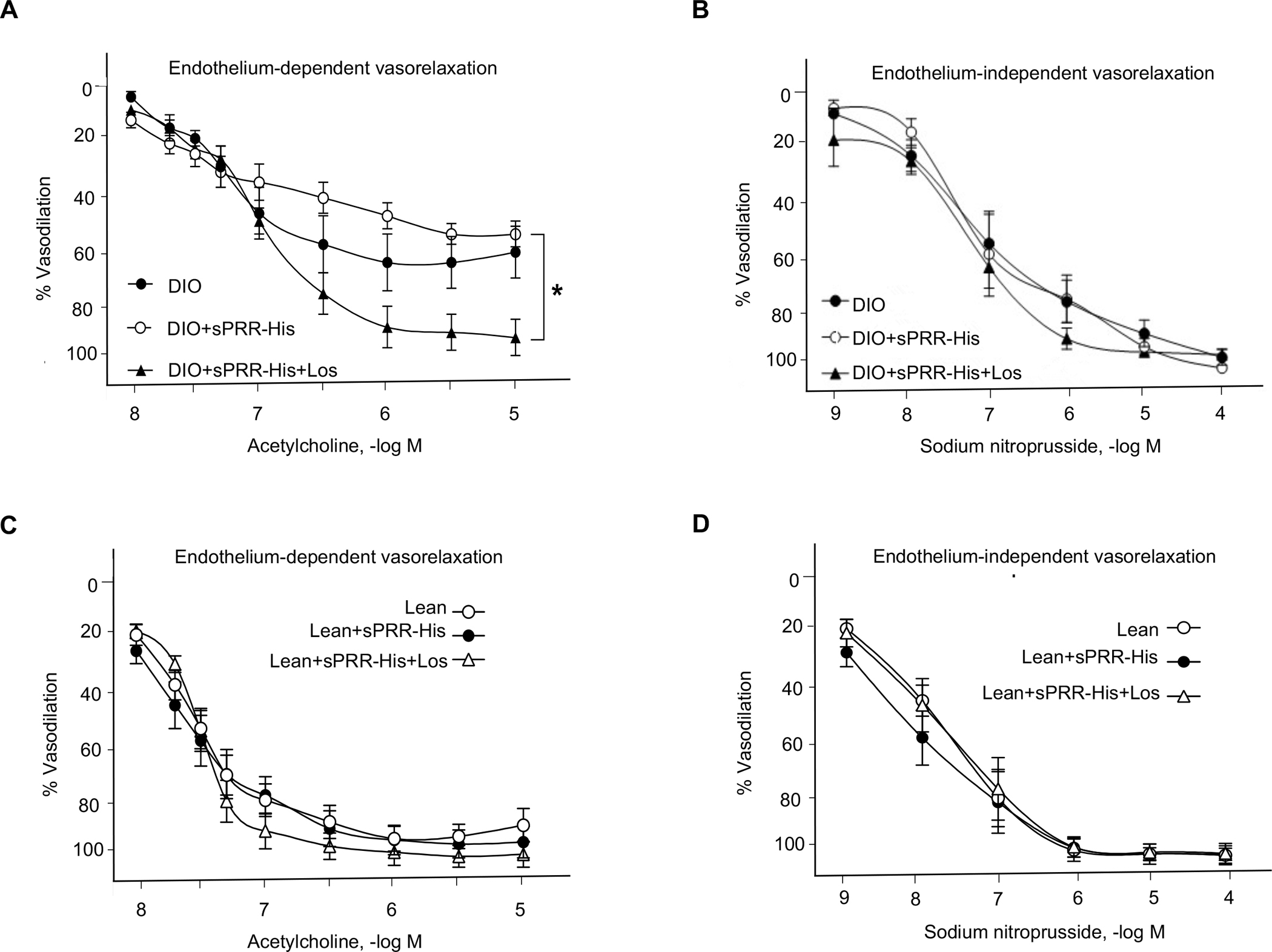
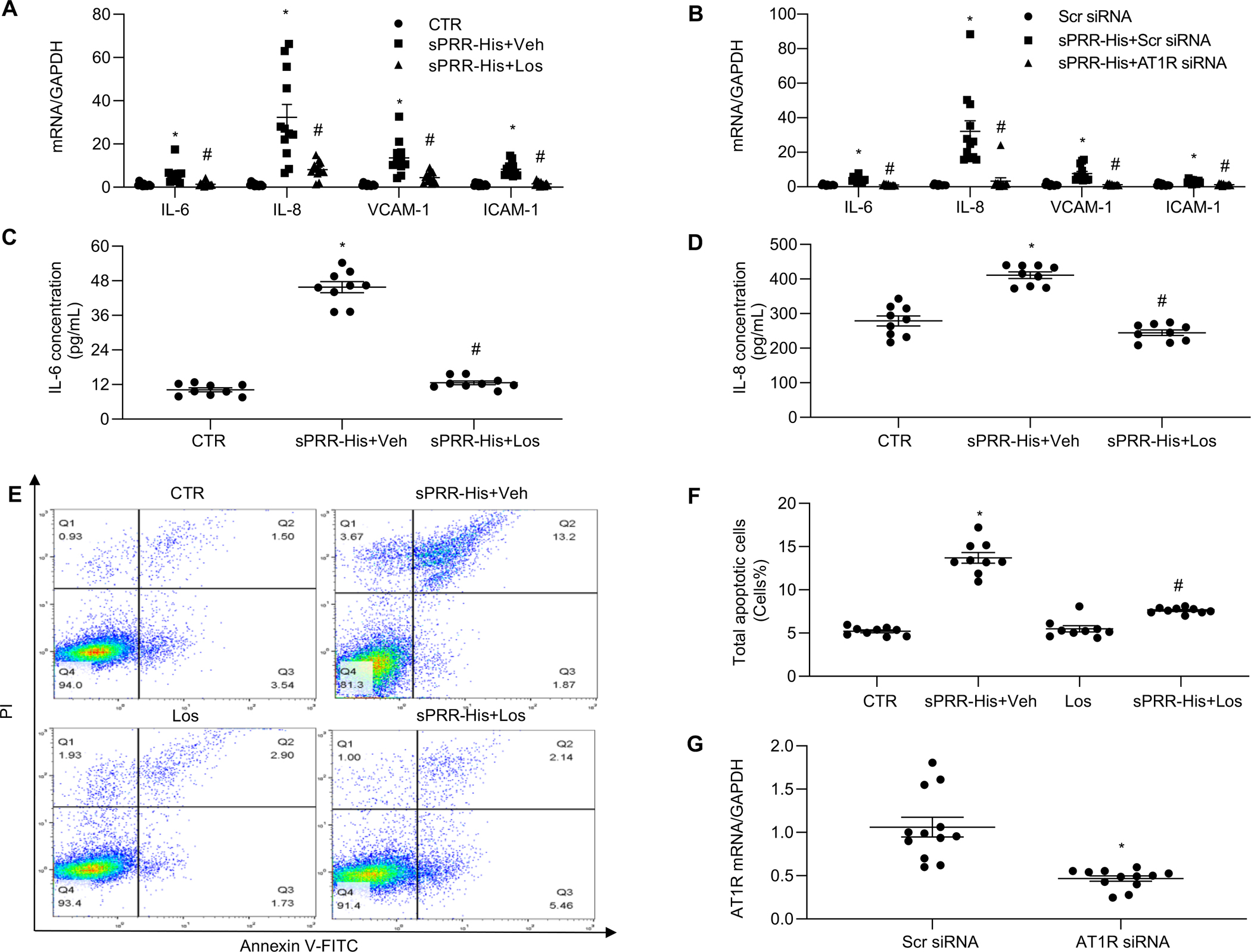
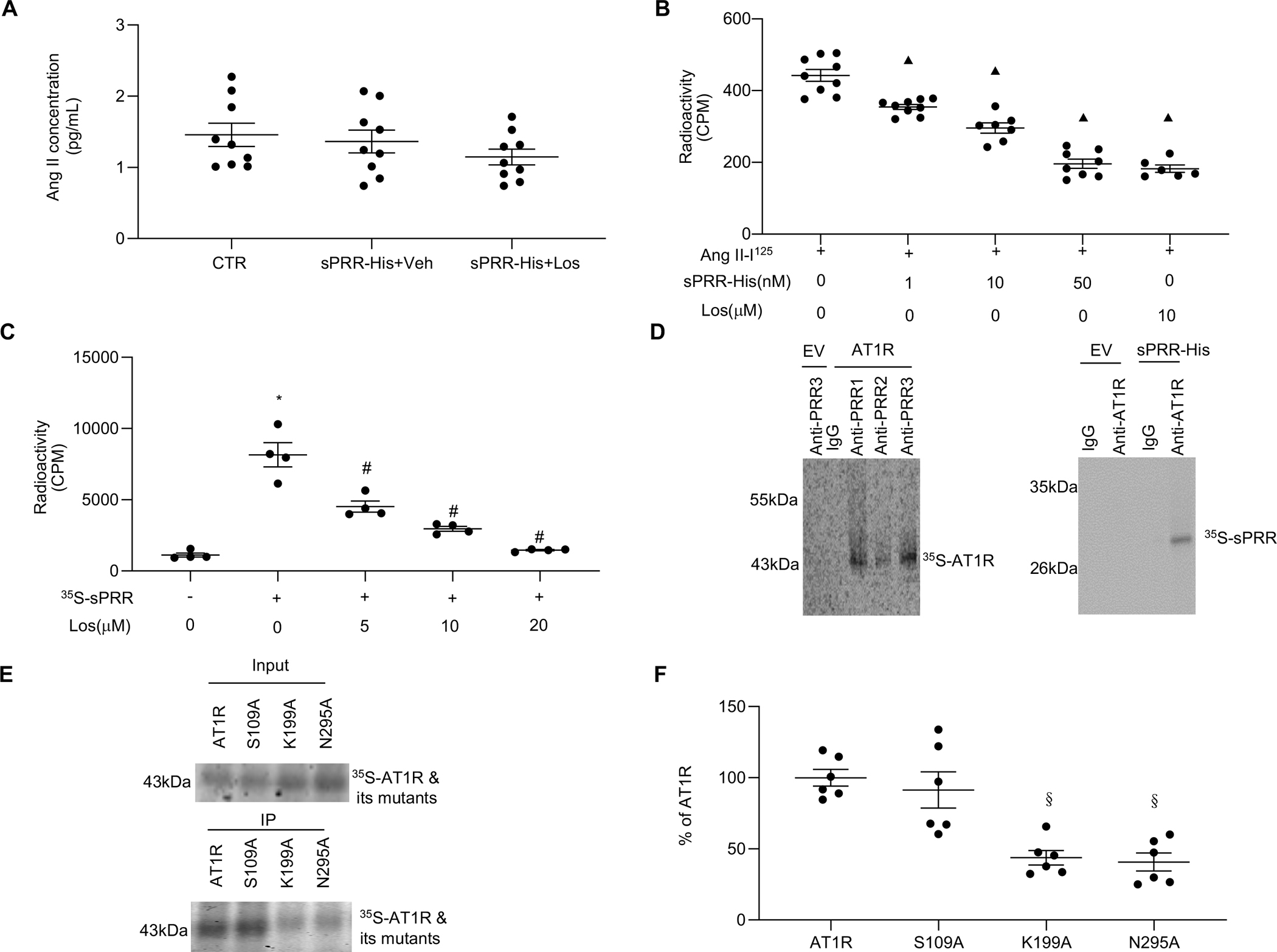
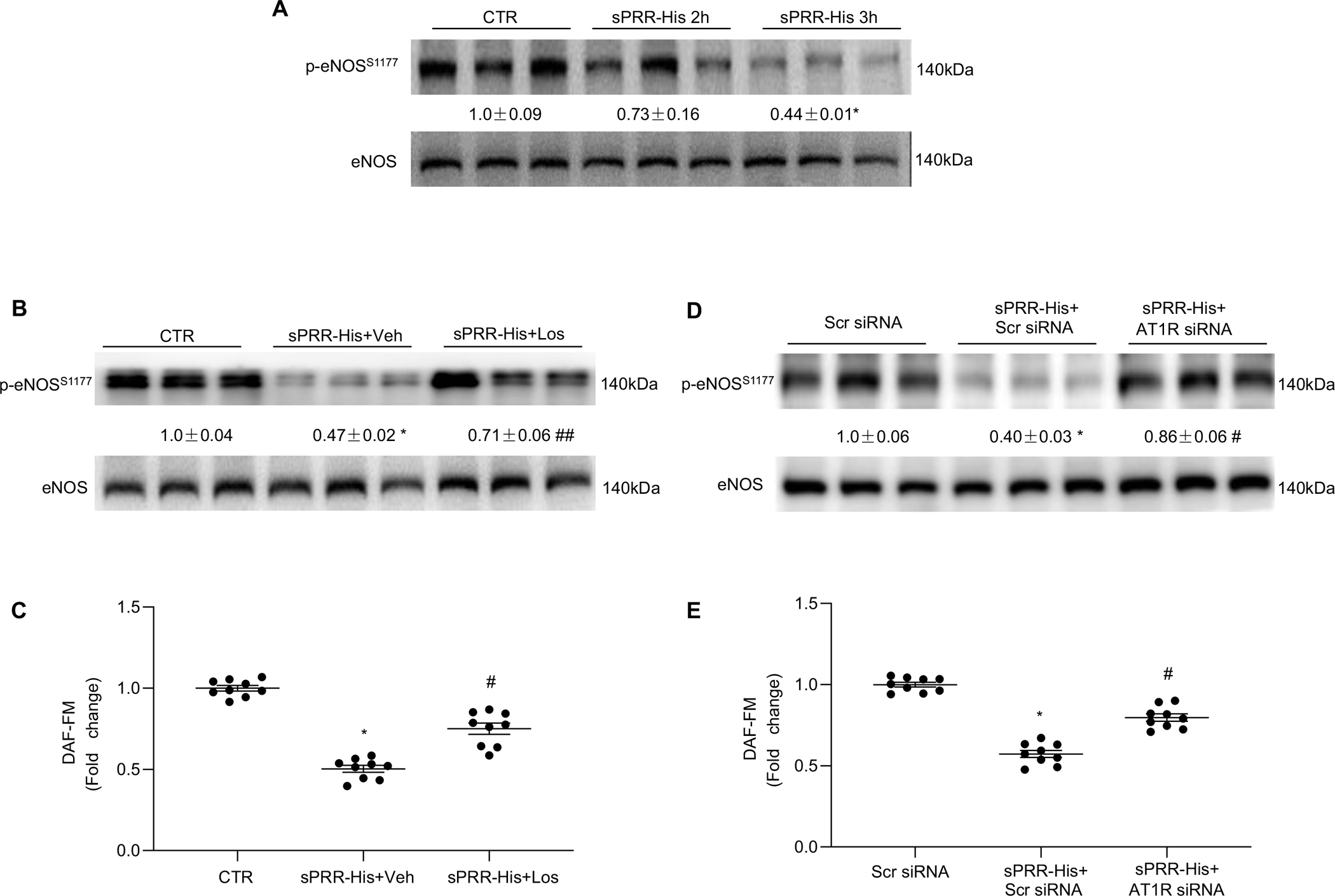
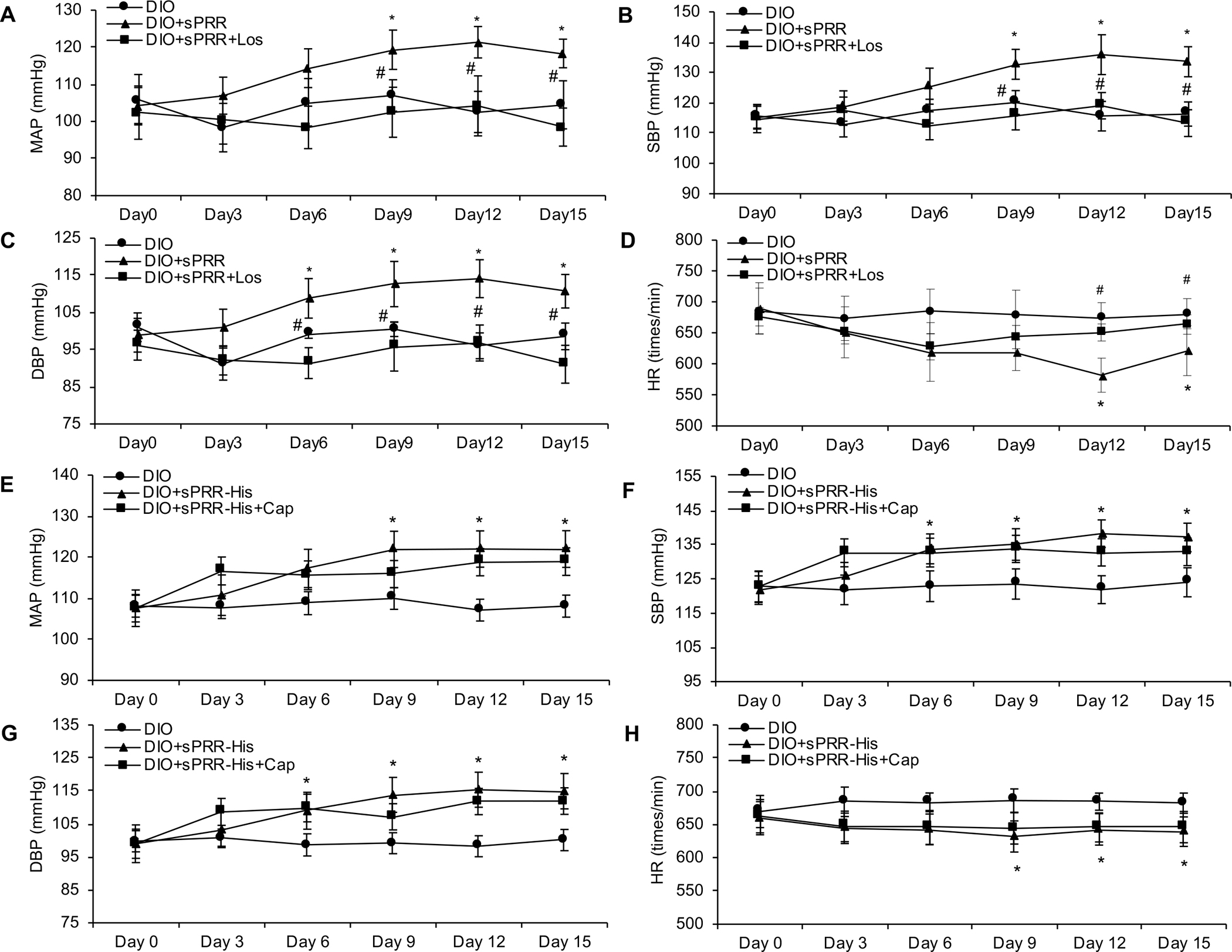
Until now, renin-angiotensin system (RAS) hyperactivity was largely thought to result from angiotensin II (Ang II)-dependent stimulation of the Ang II type 1 receptor (AT1R). Here we assessed the role of soluble (pro)renin receptor (sPRR), a product of site-1 protease-mediated cleavage of (pro)renin receptor (PRR), as a possible ligand of the AT1R in mediating: (i) endothelial cell dysfunction in vitro and (ii) arterial dysfunction in mice with diet-induced obesity. Primary human umbilical vein endothelial cells (HUVECs) treated with a recombinant histidine-tagged sPRR (sPRR-His) exhibited IκBα degradation concurrent with NF-κB p65 activation. These responses were secondary to sPRR-His evoked elevations in Nox4-derived H2O2 production that resulted in inflammation, apoptosis and reduced NO production. Each of these sPRR-His-evoked responses was attenuated by AT1R inhibition using Losartan (Los) but not ACE inhibition using captopril (Cap). Further mechanistic exploration revealed that sPRR-His activated AT1R downstream Gq signaling pathway. Immunoprecipitation coupled with autoradiography experiments and radioactive ligand competitive binding assays indicate sPRR directly interacts with AT1R via Lysine199 and Asparagine295. Important translational relevance was provided by findings from obese C57/BL6 mice that sPRR-His evoked endothelial dysfunction was sensitive to Los. Besides, sPRR-His elevated blood pressure in obese C57/BL6 mice, an effect that was reversed by concurrent treatment with Los but not Cap. Collectively, we provide solid evidence that the AT1R mediates the functions of sPRR during obesity-related hypertension. Inhibiting sPRR signaling should be considered further as a potential therapeutic intervention in the treatment and prevention of cardiovascular disorders involving elevated blood pressure.
迄今为止,肾素-血管紧张素系统(RAS)的过度活跃被认为主要是由于血管紧张素 II(Ang II)依赖性刺激血管紧张素 II 型 1 型受体(AT1R)引起的。在这里,我们评估了可溶性(前)肾素受体(sPRR)的作用,sPRR 是(前)肾素受体(PRR)经位点 1 蛋白酶切割的产物,作为可能的 AT1R 配体在介导以下方面的作用:(i)体外内皮细胞功能障碍和(ii)饮食诱导肥胖小鼠的动脉功能障碍。用重组组氨酸标记的 sPRR(sPRR-His)处理的原代人脐静脉内皮细胞(HUVEC)表现出 IκBα 降解,同时 NF-κB p65 激活。这些反应继发于 sPRR-His 引发的 Nox4 衍生的 H2O2 产生升高,导致炎症、细胞凋亡和减少 NO 产生。这些 sPRR-His 引发的反应都被使用 Losartan(Los)的 AT1R 抑制减弱,但使用 Captopril(Cap)的 ACE 抑制则不然。进一步的机制探索表明,sPRR-His 激活了 AT1R 下游的 Gq 信号通路。免疫沉淀结合放射自显影实验和放射性配体竞争结合实验表明,sPRR 通过赖氨酸 199 和天冬酰胺 295 直接与 AT1R 相互作用。肥胖 C57/BL6 小鼠的发现提供了重要的转化相关性,即 sPRR-His 引起的内皮功能障碍对 Los 敏感。此外,sPRR-His 升高肥胖 C57/BL6 小鼠的血压,这种效应可被同时给予 Los 但不是 Cap 逆转。总的来说,我们提供了确凿的证据表明,在肥胖相关高血压中,AT1R 介导了 sPRR 的功能。抑制 sPRR 信号应该被进一步考虑作为一种潜在的治疗干预,以治疗和预防涉及血压升高的心血管疾病。