Torrado Mario, Fernández Germán, Ganoza Christian A, Maneiro Emilia, García Diego, Sonicheva-Paterson Natalia, Rosa Isaac, Ochoa Juan Pablo, Santomé Luis, Vasichkina Elena, Monserrat Lorenzo
Institute of Health Sciences, University of A Coruña, A Coruña, Spain.
Cardiovascular Genetics, Health in Code, A Coruña, Spain.
NPJ Genom Med. 2021 Mar 4;6(1):21. doi: 10.1038/s41525-021-00183-y.
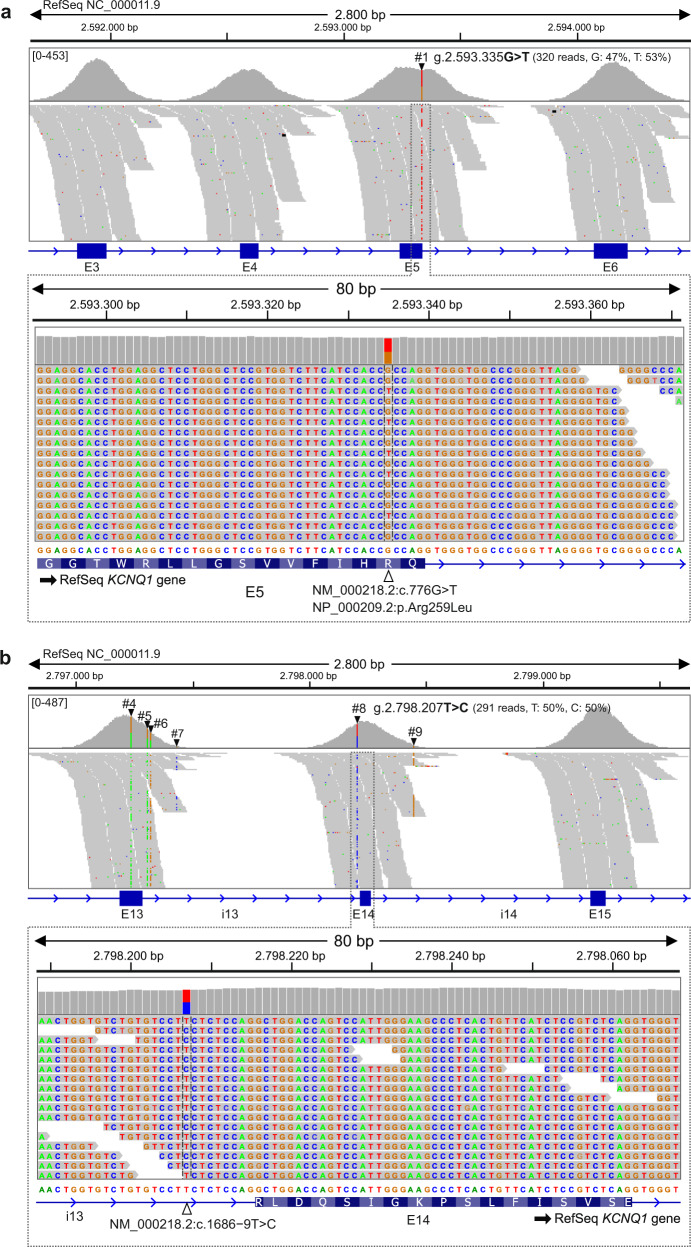
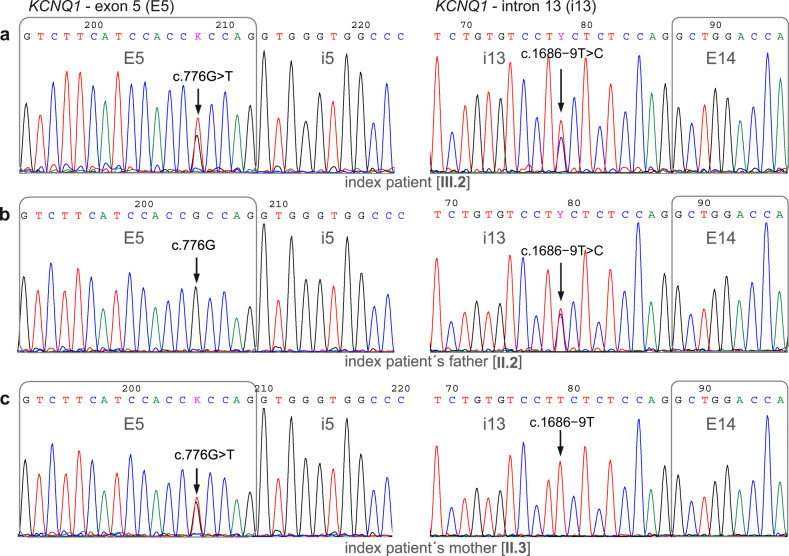
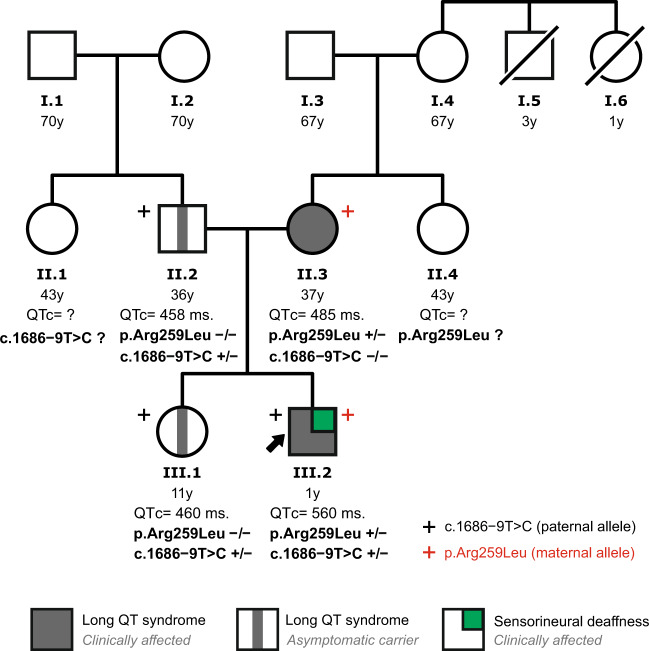
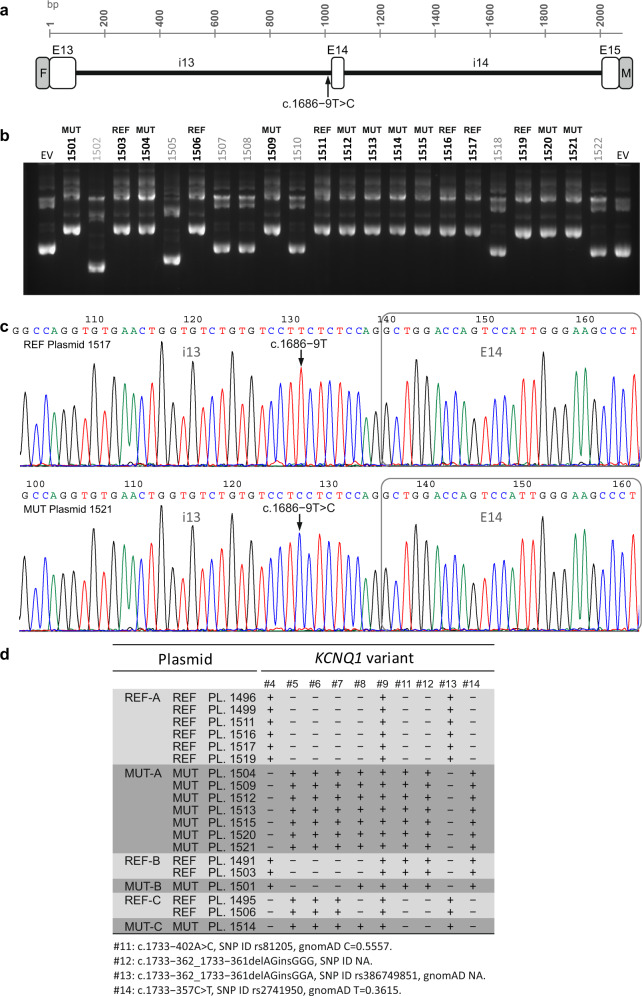
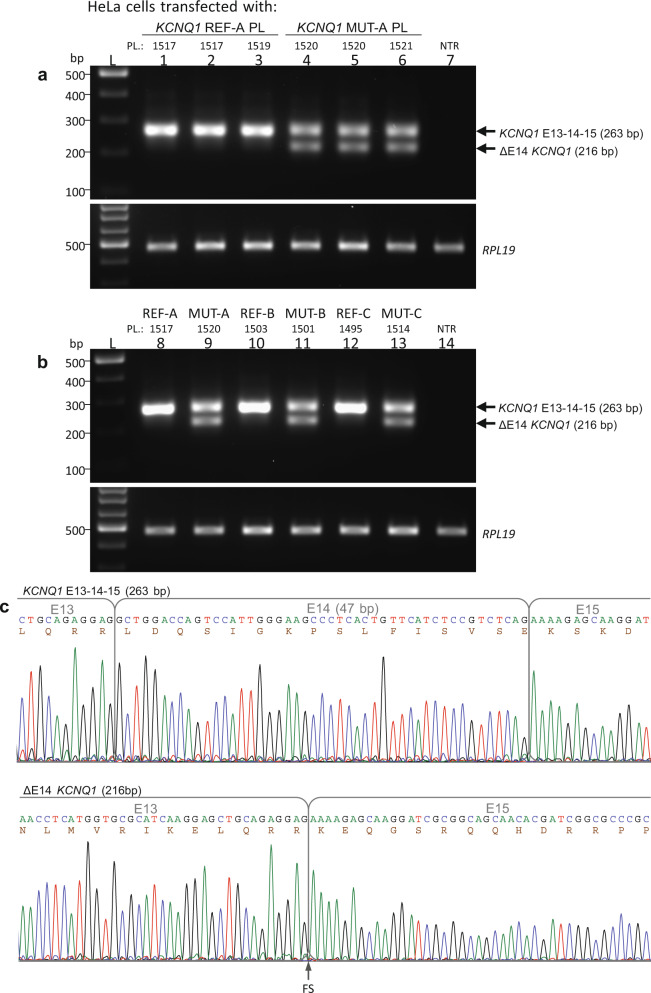
Here we report an infant with clinical findings suggestive of Jervell and Lange-Nielsen syndrome (JLNS), including a prolonged QT interval (LQTS) and chronic bilateral sensorineural deafness. NGS analysis revealed one known heterozygous pathogenic missense variant, KCNQ1 p.R259L, previously associated with LQTS but insufficient to explain the cardioauditory disorder. In a screening of proximal intronic regions, we found a heterozygous variant, KCNQ1 c.1686-9 T > C, absent from controls and previously undescribed. Several splicing prediction tools returned low scores for this intronic variant. Driven by the proband's phenotype rather than the neutral predictions, we have characterized this rare intronic variant. Family analysis has shown that the proband inherited the missense and the intronic variants from his mother and father, respectively. A minigene splicing assay revealed that the intronic variant induced an additional transcript, arising from skipping of exon 14, which was translated into a truncated protein in transfected cells. The splice-out of exon 14 creates a frameshift in exon 15 and a stop codon in exon 16, which is the last exon of KCNQ1. This mis-spliced transcript is expected to escape nonsense-mediated decay and predicted to encode a truncated loss-of-function protein, KCNQ1 p.L563Kfs*73. The analysis of endogenous KCNQ1 expression in the blood of the proband's parents detected the aberrant transcript only in the patient's father. Taken together, these analyses confirmed the proband's diagnosis of JLNS1 and indicated that c.1686-9 T > C is a cryptic splice-altering variant, expanding the known genetic spectrum of biallelic KCNQ1 variant combinations leading to JLNS1.
在此,我们报告一名婴儿,其临床表现提示为杰韦尔和朗格 - 尼尔森综合征(JLNS),包括QT间期延长(LQTS)和慢性双侧感音神经性耳聋。二代测序(NGS)分析发现一个已知的杂合致病性错义变异,KCNQ1 p.R259L,此前与LQTS相关,但不足以解释心脏听觉障碍。在对近端内含子区域的筛查中,我们发现了一个杂合变异,KCNQ1 c.1686 - 9 T > C,对照组中未出现且此前未被描述。几种剪接预测工具对该内含子变异给出的分数较低。受先证者表型而非中性预测结果的驱动,我们对这个罕见的内含子变异进行了特征分析。家系分析表明,先证者分别从其母亲和父亲那里继承了错义变异和内含子变异。一个小基因剪接分析显示,该内含子变异诱导产生了一种额外的转录本,它是由于外显子14的跳跃产生的,在转染细胞中被翻译成一种截短蛋白。外显子14的缺失在第15外显子中产生移码,并在第16外显子(即KCNQ1的最后一个外显子)中产生一个终止密码子。这种错误剪接的转录本预计会逃避无义介导的衰变,并预计编码一种截短的功能丧失蛋白,KCNQ1 p.L563Kfs*73。对先证者父母血液中内源性KCNQ1表达的分析仅在患者父亲中检测到了异常转录本。综上所述,这些分析证实了先证者的JLNS1诊断,并表明c.1686 - 9 T > C是一个隐匿的剪接改变变异,扩展了导致JLNS1的双等位基因KCNQ1变异组合的已知遗传谱。