Center for Neurogenetics, Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY, USA.
Neurogenetics Research, Weill Cornell Medicine Qatar, Doha, Qatar.
Genet Med. 2021 Jul;23(7):1211-1218. doi: 10.1038/s41436-021-01126-9. Epub 2021 Mar 8.
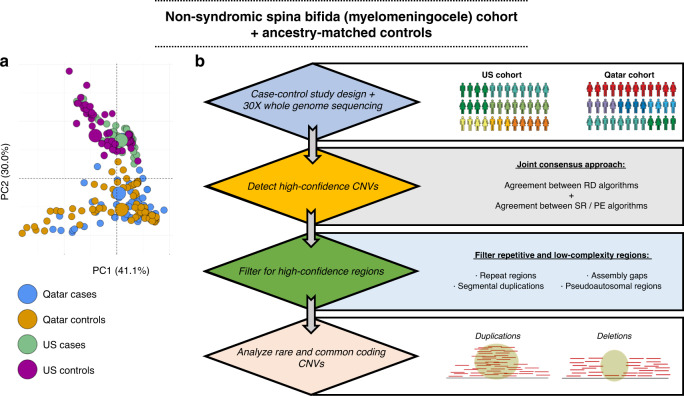
Next-generation sequencing has implicated some risk variants for human spina bifida (SB), but the genome-wide contribution of structural variation to this complex genetic disorder remains largely unknown. We examined copy-number variant (CNV) participation in the genetic architecture underlying SB risk.
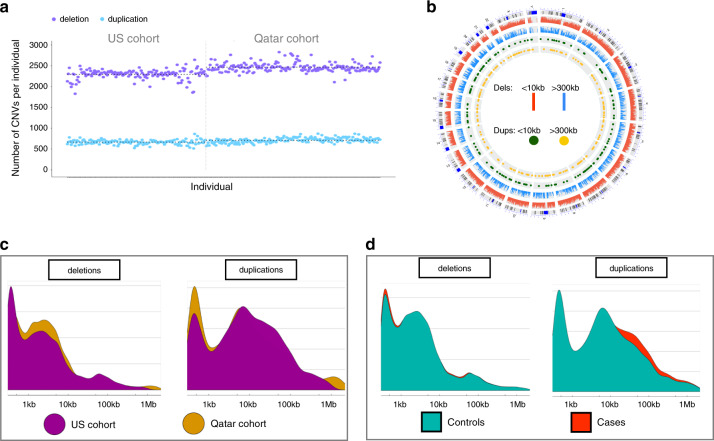
A high-confidence ensemble approach to genome sequences (GS) was benchmarked and employed for systematic detection of common and rare CNVs in two separate ancestry-matched SB case-control cohorts.
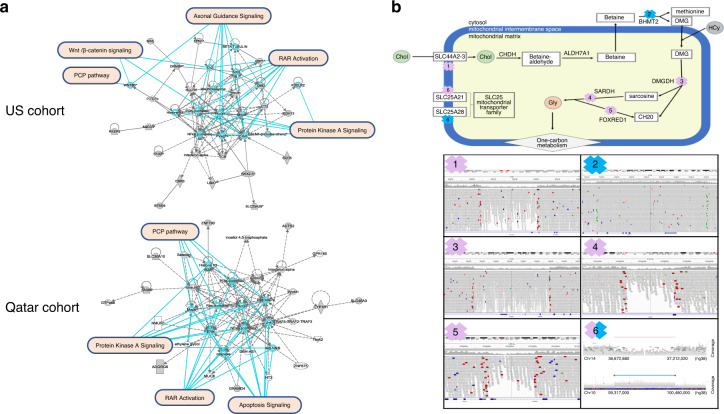
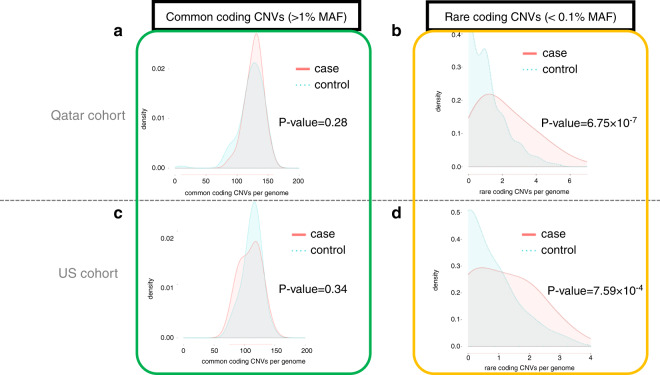
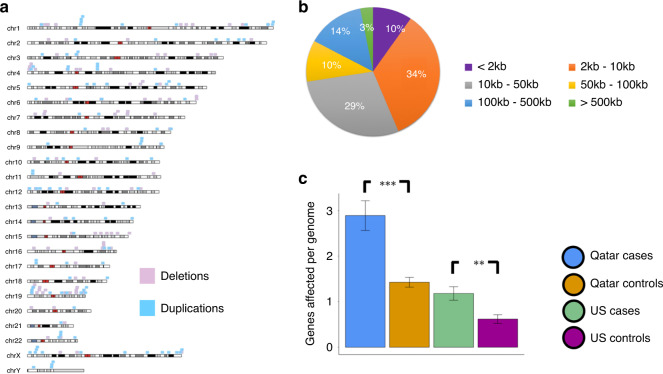
SB cases were enriched with exon disruptive rare CNVs, 44% of which were under 10 kb, in both ancestral populations (P = 6.75 × 10; P = 7.59 × 10). Genes containing these disruptive CNVs fall into molecular pathways, supporting a role for these genes in SB. Our results expand the catalog of variants and genes with potential contribution to genetic and gene-environment interactions that interfere with neurulation, useful for further functional characterization.
This study underscores the need for genome-wide investigation and extends our previous threshold model of exonic, single-nucleotide variation toward human SB risk to include structural variation. Since GS data afford detection of CNVs with greater resolution than microarray methods, our results have important implications toward a more comprehensive understanding of the genetic risk and mechanisms underlying neural tube defect pathogenesis.
下一代测序技术已经发现了一些人类脊柱裂(SB)的风险变异,但结构变异对这种复杂遗传疾病的全基因组贡献在很大程度上仍不清楚。我们研究了拷贝数变异(CNV)在 SB 风险遗传结构中的作用。
采用高可信度的基因组序列(GS)综合方法,对两个具有相似祖先的 SB 病例对照队列进行了常见和罕见 CNV 的系统检测。
SB 病例在两个祖先群体中均富集了exon 破坏的罕见 CNV,其中 44%的长度小于 10kb(P=6.75×10;P=7.59×10)。包含这些破坏 CNV 的基因属于分子途径,支持这些基因在 SB 中的作用。我们的结果扩展了可能导致基因-环境相互作用干扰神经发生的变异和基因目录,有助于进一步的功能特征描述。
本研究强调了全基因组研究的必要性,并将我们之前关于exon 单核苷酸变异对人类 SB 风险的外显子阈值模型扩展到包括结构变异。由于 GS 数据可以比微阵列方法更精确地检测 CNV,因此我们的结果对于更全面地理解神经管缺陷发病机制中的遗传风险和机制具有重要意义。