Feng Li, Wang Songhua, Chen Feng, Zhang Cheng, Wang Qiao, Zhao Yuting, Zhang Zifeng
School of Life Science, Jiangsu Normal University, Xuzhou, Jiangsu, 221116, People's Republic of China.
Diabetes Metab Syndr Obes. 2021 Mar 2;14:963-981. doi: 10.2147/DMSO.S299570. eCollection 2021.
Emerging evidence from animal studies and clinical trials indicates that systemic inhibition of endothelin1 (ET1) signaling by endothelin receptor antagonists improves pathological features of diabetes and its complications. It is indicated that endothelin type A receptor (ETAR) plays a major role in ET1-mediated pathophysiological actions including diabetic pathology. However, the effects as well as the mechanistic targets of hepatic ET1/ETAR signaling inhibition on the pathology of metabolic diseases remain unclear. This study aimed to investigate the beneficial effects as well as the underlying mechanisms of hepatic ETAR knockdown on metabolism abnormalities in high-fat diet (HFD)-fed mice.
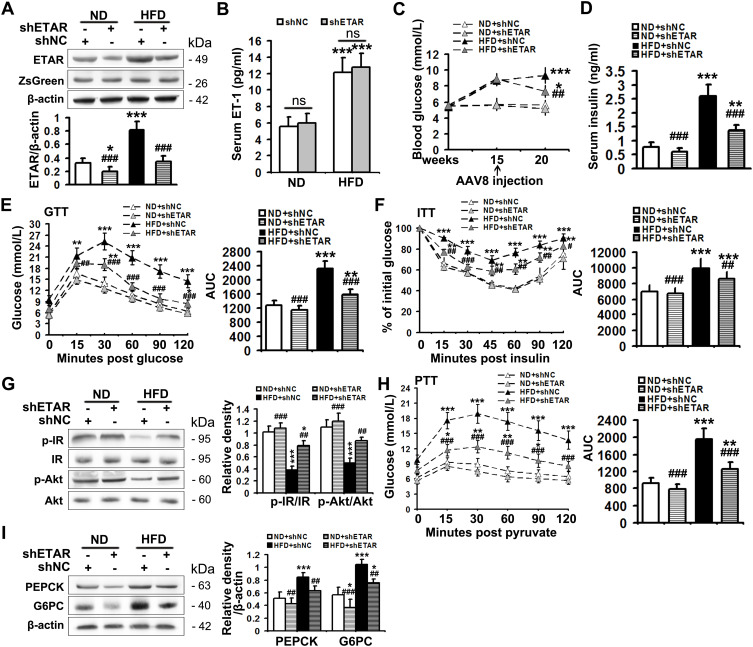
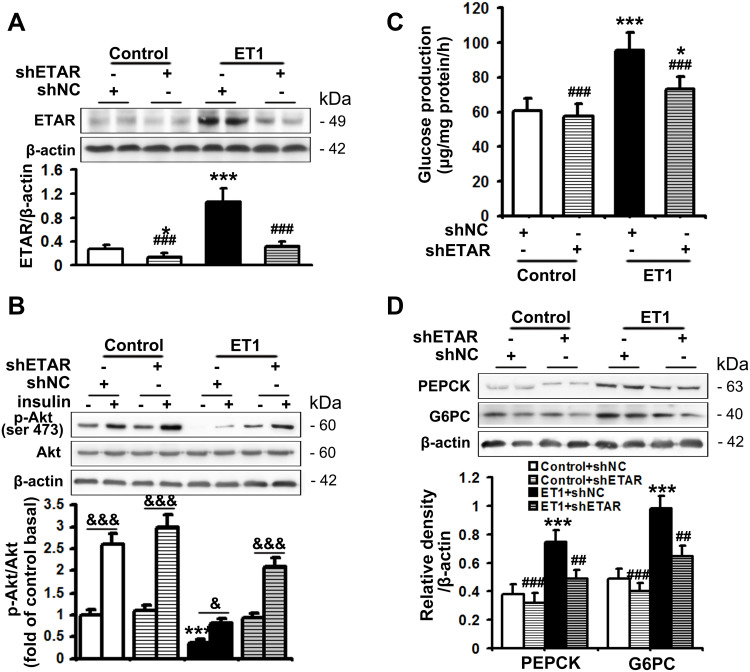
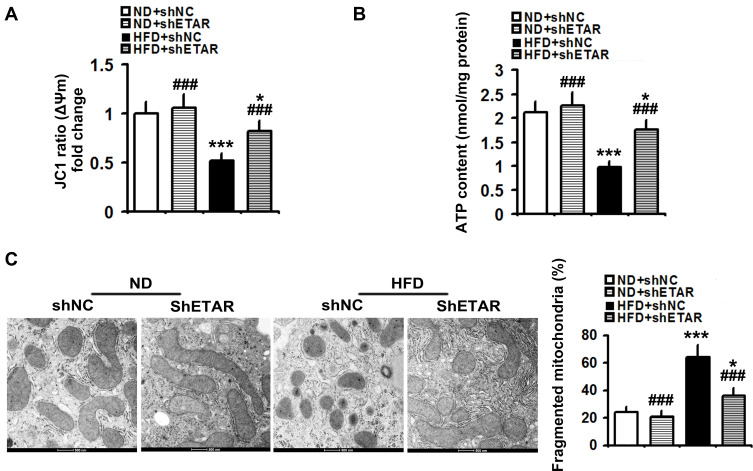
Mice were fed a HFD to induce insulin resistance and metabolism abnormalities. L02 cells were treated with ET1 to assess the action of ET1/ETAR signaling in vitro. Liver-selective knockdown of ETAR was achieved by tail vein injection of adeno-associated virus 8 (AAV8). Systemic and peripheral metabolism abnormalities were determined in vivo and in vitro. Mitochondrial fragmentation was observed by transmission electron microscope (TEM) and mitoTracker red staining.
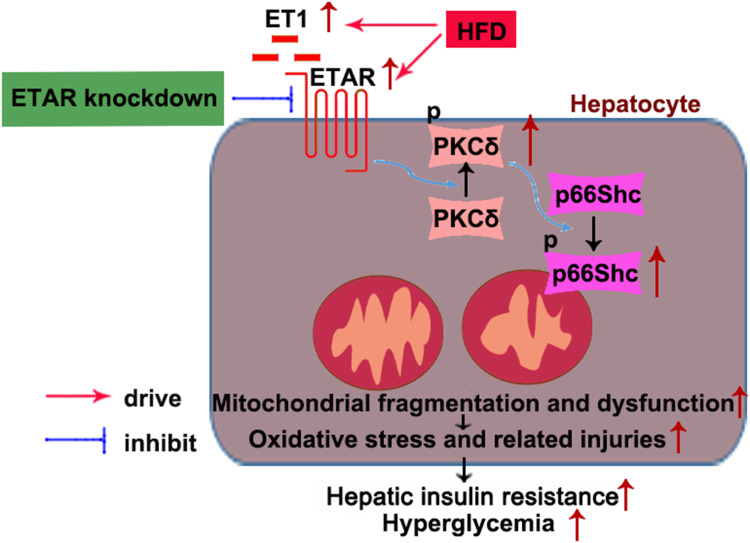
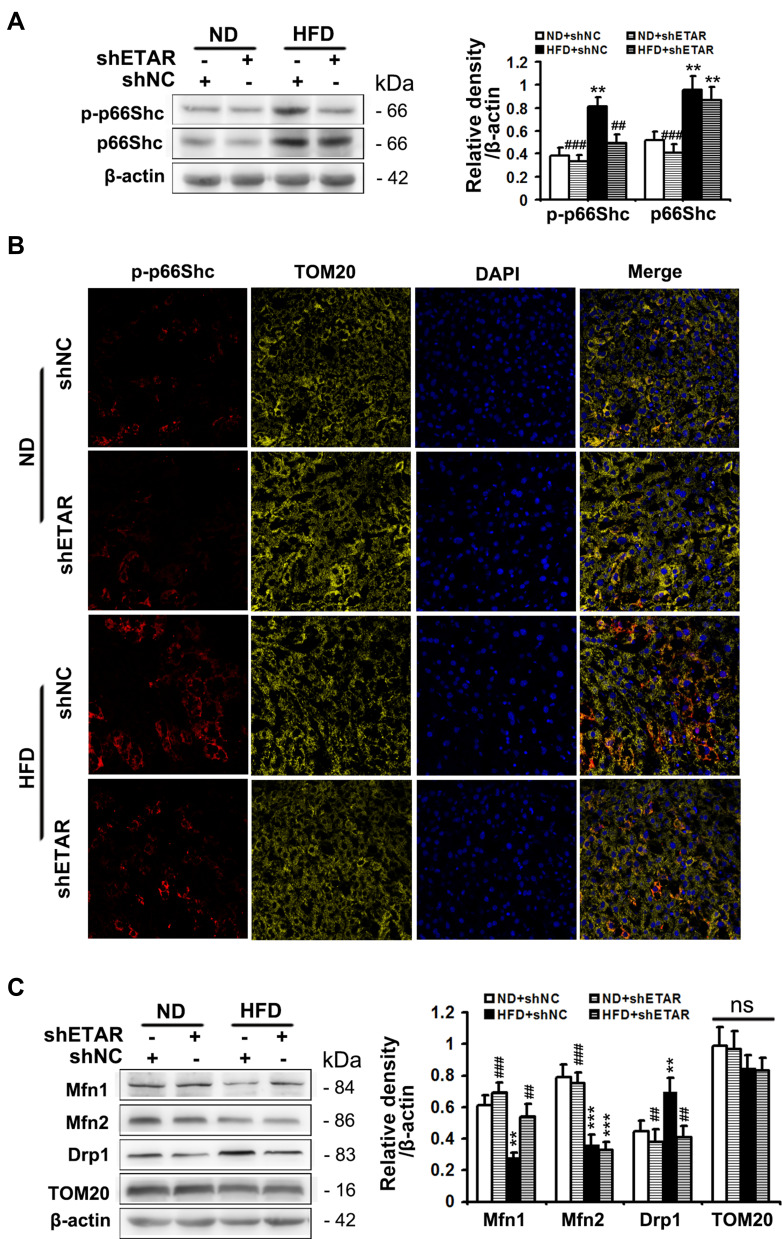
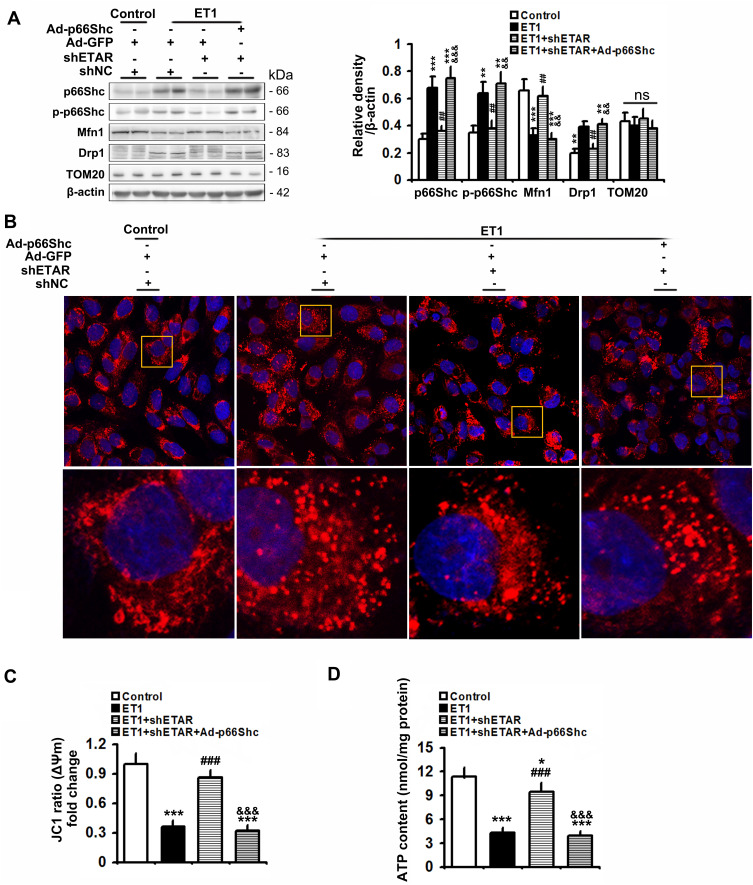
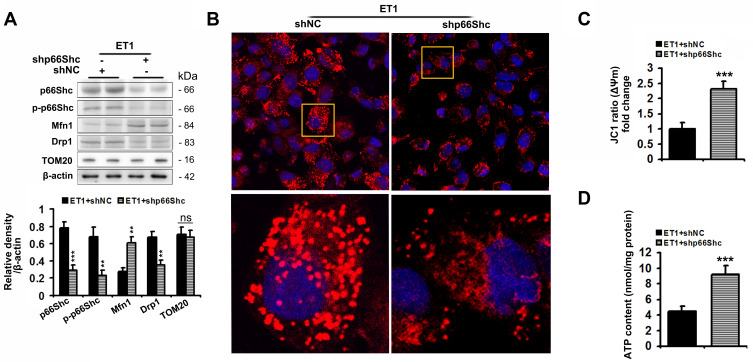
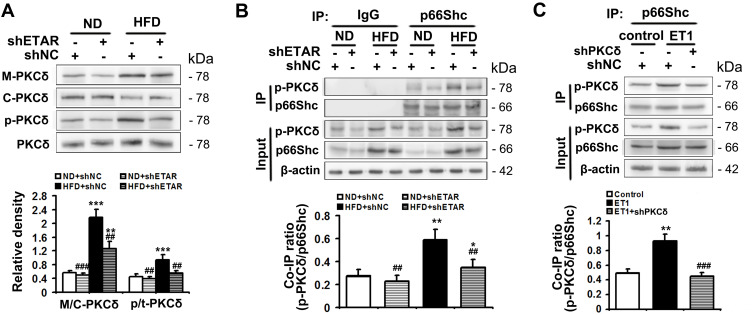
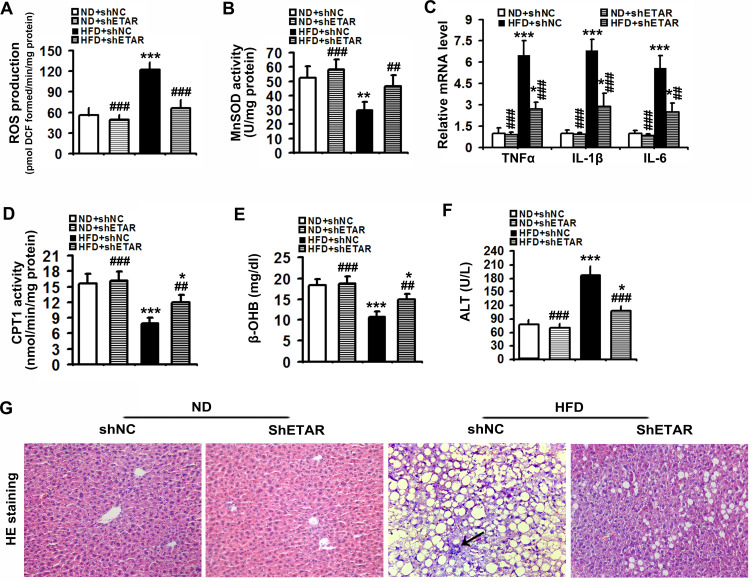
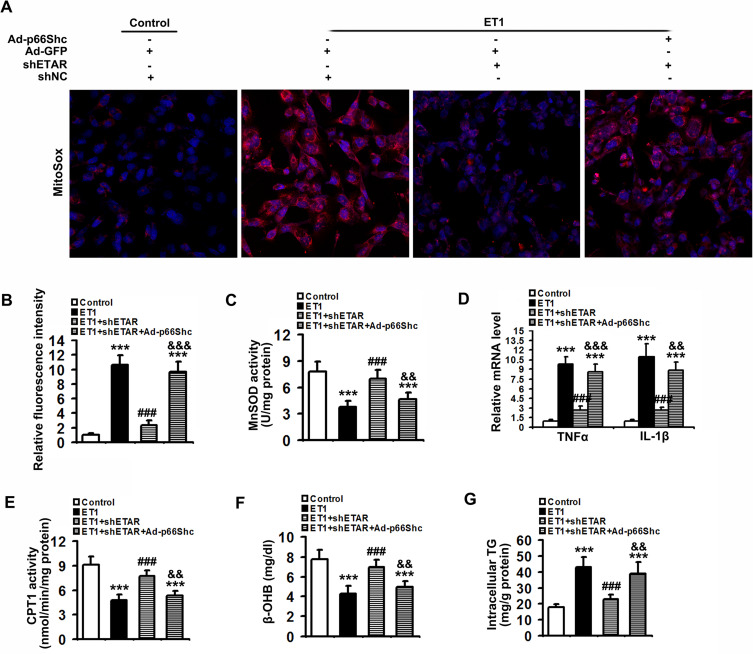
Here we provided in vivo and in vitro evidence to demonstrate that liver-selective knockdown of ETAR effectively ameliorated hepatic insulin resistance and hyperglycemia in HFD-fed mice. Mechanistically, hepatic ETAR knockdown alleviated mitochondrial fragmentation and dysfunction via inactivating 66-kDa Src homology 2 domain-containing protein (p66Shc) to recover mitochondrial dynamics, which was mediated by inhibiting protein kinase Cδ (PKCδ), in the livers of HFD-fed mice. Ultimately, hepatic ETAR knockdown attenuated mitochondria-derived oxidative stress and related liver injuries in HFD-fed mice. These ETAR knockdown-mediated actions were confirmed in ET1-treated L02 cells.
This study defined an ameliorative role of hepatic ETAR knockdown in HFD-induced metabolism abnormalities by alleviating p66Shc-mediated mitochondrial fragmentation and consequent oxidative stress-related disorders and indicated that hepatic ETAR knockdown may be a promising therapeutic strategy for metabolic diseases.
动物研究和临床试验的新证据表明,内皮素受体拮抗剂对内皮素1(ET1)信号通路的全身抑制作用可改善糖尿病及其并发症的病理特征。研究表明,A型内皮素受体(ETAR)在ET1介导的病理生理作用(包括糖尿病病理)中起主要作用。然而,肝脏ET1/ETAR信号通路抑制对代谢性疾病病理的影响及其作用机制靶点仍不清楚。本研究旨在探讨肝脏ETAR基因敲低对高脂饮食(HFD)喂养小鼠代谢异常的有益作用及其潜在机制。
给小鼠喂食HFD以诱导胰岛素抵抗和代谢异常。用ET1处理L02细胞以评估ET1/ETAR信号通路在体外的作用。通过尾静脉注射腺相关病毒8(AAV8)实现肝脏选择性敲低ETAR。在体内和体外测定全身和外周代谢异常。通过透射电子显微镜(TEM)和线粒体红色荧光探针染色观察线粒体碎片化。
在此,我们提供了体内和体外证据,证明肝脏选择性敲低ETAR可有效改善HFD喂养小鼠的肝脏胰岛素抵抗和高血糖。机制上,肝脏ETAR基因敲低通过使含66-kDa Src同源结构域蛋白(p66Shc)失活来减轻线粒体碎片化和功能障碍,从而恢复线粒体动力学,这是通过抑制蛋白激酶Cδ(PKCδ)介导的,在HFD喂养小鼠的肝脏中。最终,肝脏ETAR基因敲低减轻了HFD喂养小鼠线粒体衍生的氧化应激和相关肝损伤。这些ETAR基因敲低介导的作用在ET1处理的L02细胞中得到证实。
本研究通过减轻p66Shc介导的线粒体碎片化及随之而来的氧化应激相关紊乱,确定了肝脏ETAR基因敲低在HFD诱导的代谢异常中的改善作用,并表明肝脏ETAR基因敲低可能是治疗代谢性疾病的一种有前景的治疗策略。