Systemic Autoimmunity Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Institutes of Health, Bethesda, MD 20892, USA.
Science. 2021 Mar 12;371(6534):1100-1101. doi: 10.1126/science.abg6449.
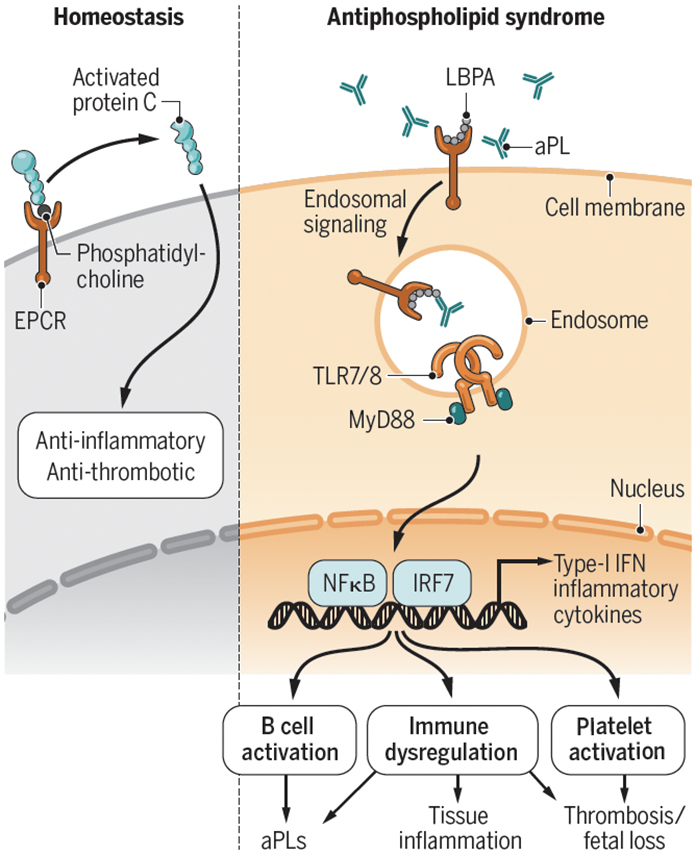
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by increased risk for arterial, venous and/or microvascular thrombosis and various obstetrical complications, including recurrent miscarriages, premature births, and preeclampsia, in association with the persistent presence of phospholipid antibodies (aPLs). The clinical spectrum of APS ranges from mild clinical manifestations to the development of a catastrophic event involving multiorgan failure and high mortality due to disseminated thrombosis. APS diagnosis requires the detection of serum autoantibodies targeting cardiolipins and/or the plasma protein β-2-glycoprotein I (β2GPI). However, the full spectrum of specific autoantigens primarily recognized by aPLs remain unknown, which has hampered therapeutic development. On page XXX of this issue, Muller-Calleja (1) report the identification of a cell surface antigenic complex composed of endosomal lysobiphosphatidic acid (LBPA) presented by the endothelial protein C receptor (EPCR), which is specifically recognized by aPLs and promotes immune dysregulation and thrombosis in mice.
抗磷脂综合征(APS)是一种以动脉、静脉和/或微血管血栓形成风险增加以及与持续存在磷脂抗体(aPL)相关的各种产科并发症为特征的自身免疫性疾病,包括复发性流产、早产和子痫前期。APS 的临床谱范围从轻微的临床表现到涉及多器官衰竭和高死亡率的灾难性事件的发展,这是由于弥散性血栓形成。APS 的诊断需要检测针对心磷脂和/或血浆蛋白 β-2-糖蛋白 I(β2GPI)的血清自身抗体。然而,aPLs 主要识别的特定自身抗原的全貌仍不清楚,这阻碍了治疗的发展。在本期第 XXX 页,Muller-Calleja (1)报道了一种由内体溶血磷脂酸(LBPA)组成的细胞表面抗原复合物的鉴定,该复合物由内皮蛋白 C 受体(EPCR)呈递,该复合物被 aPLs 特异性识别,并促进小鼠的免疫失调和血栓形成。