Foran Jason, Moore Michael, Crushell Ellen, Knerr Ina, McSweeney Niamh
Department of Paediatric Neurology Cork University Hospital Cork Republic of Ireland.
Department of Radiology Cork University Hospital Cork Republic of Ireland.
JIMD Rep. 2020 Nov 16;58(1):12-20. doi: 10.1002/jmd2.12187. eCollection 2021 Mar.
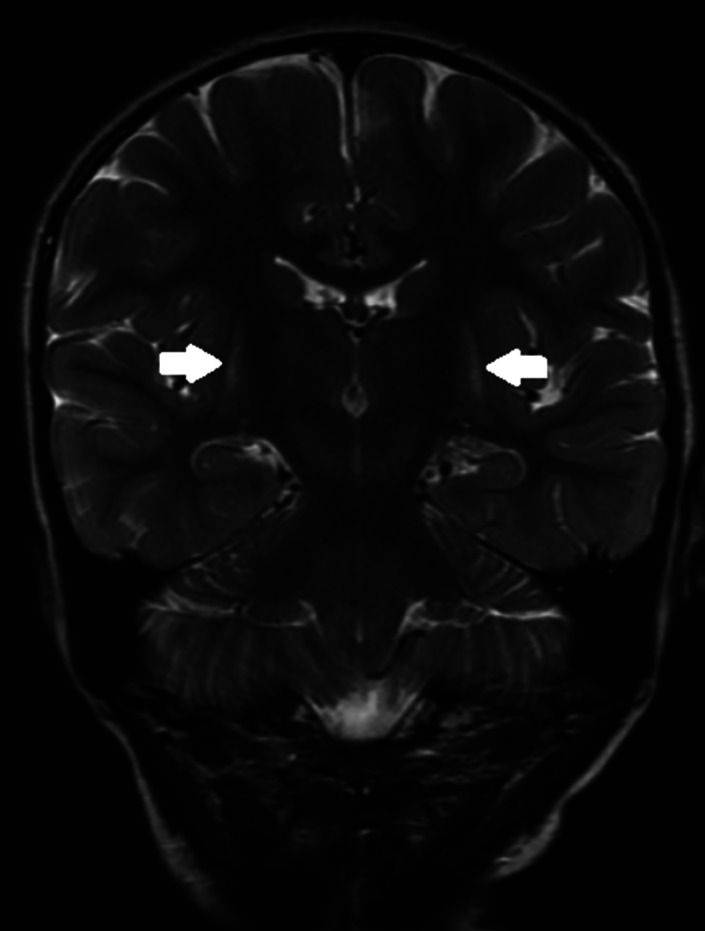
A 4-year-old girl was referred for reassessment of dyskinetic cerebral palsy. Initial investigations in her country of birth, India, had not yielded a diagnosis. MRI brain in infancy revealed bilateral putamen hyperintensity. She had generalized dyskinesia predominantly bulbar and limbs. Motor and speech development were most affected with preservation of cognitive development. There was no history of acute encephalopathic crisis or status dystonicus. Initial urine organic acids and amino acids and acylcarnitine profile (ACP) were normal. A dystonia genetic panel showed compound heterozygosity with a pathogenic variant and a variant of uncertain significance in the gene. The latter is hitherto undescribed and is indicative of a potential diagnosis of glutaric aciduria type 1 (alternatively glutaric acidemia type 1) (GA-1), an autosomal recessive disorder of mitochondrial lysine/hydroxylysine and tryptophan metabolism. Repeat urine organic acids showed isolated slightly increased 3-hydroxy glutarate excretion consistent with GA-1 and characterizing the patient as a "low excretor," a diagnostic sub-group where diagnosis is more challenging but prognosis is similar. Repeat MRI Brain at age 4 showed volume loss and symmetric T2 hyperintensity in the posterior putamina bilaterally. This case highlights the diagnostic dilemma of GA-1 where differing clinical courses, genetic variants, neuroradiological findings, and biochemical excretion patterns may lead to a later diagnosis. The presence of newborn screening for GA-1 should not dull the clinician's suspicion of the possibility that GA-1 may present with a complex movement disorder. Timely diagnosis and treatment is essential, as neurological sequelae are largely irreversible.
一名4岁女童因运动障碍型脑瘫接受重新评估。她出生于印度,在当地进行的初步检查未能明确诊断。婴儿期的脑部MRI显示双侧壳核高信号。她有全身性运动障碍,主要累及延髓和四肢。运动和语言发育受影响最大,认知发育保留。无急性脑病危机或肌张力障碍状态病史。初始尿有机酸、氨基酸和酰基肉碱谱(ACP)正常。肌张力障碍基因检测显示为复合杂合子,一个致病变异和该基因中一个意义未明的变异。后者此前未被描述,提示可能诊断为1型戊二酸尿症(又称1型戊二酸血症)(GA - 1),这是一种线粒体赖氨酸/羟赖氨酸和色氨酸代谢的常染色体隐性疾病。重复检测尿有机酸显示3 - 羟基戊二酸排泄单独轻度增加,与GA - 1相符,该患者属于“低排泄者”这一诊断亚组,在此亚组中诊断更具挑战性,但预后相似。4岁时重复脑部MRI显示双侧壳核后部体积减小和对称T2高信号。该病例突出了GA - 1的诊断困境,不同的临床病程、基因变异、神经放射学表现和生化排泄模式可能导致诊断延迟。GA - 1新生儿筛查的存在不应削弱临床医生对GA - 1可能表现为复杂运动障碍的怀疑。及时诊断和治疗至关重要,因为神经后遗症在很大程度上是不可逆的。