Chen Qing-Lian, Yan Qian, Feng Kun-Liang, Xie Chun-Feng, Fang Chong-Kai, Wang Ji-Nan, Liu Li-Hua, Li Ya, Zhong Chong
Guangzhou University of Chinese Medicine, Guangzhou, 510405, People's Republic of China.
Department of Hepatobiliary Surgery, The First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, 510405, People's Republic of China.
Int J Gen Med. 2021 Mar 10;14:805-823. doi: 10.2147/IJGM.S294505. eCollection 2021.
For the identification of abnormally methylated differentially expressed genes (MDEGs) in hepatocellular carcinoma (HCC), this study integrated four microarray datasets to investigate the fundamental mechanisms of tumorigenesis.
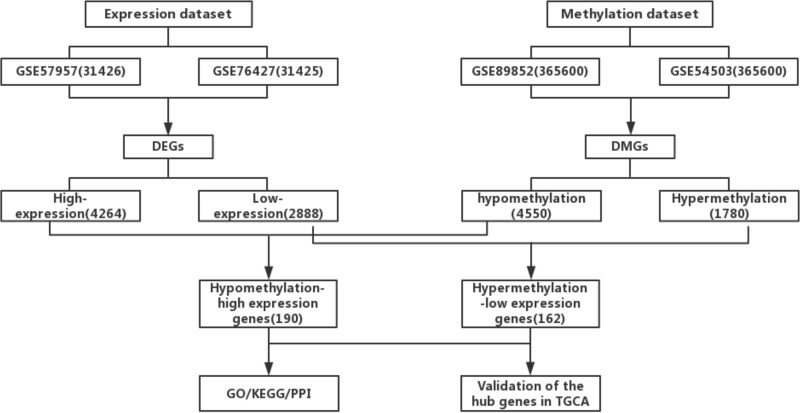
We obtained the expression (GSE76427, GSE57957) and methylation (GSE89852, GSE54503) profiles from Gene Expression Omnibus (GEO). The abnormally MDEGs were identified by using R software. We used the clusterProfiler package for the functional and pathway enrichment analysis. The String database was used to build the protein-protein interaction (PPI) network and visualize it in Cytoscape. MCODE was employed in the module analysis. Additionally, Gene Expression Profiling Interactive Analysis (GEPIA) and The Cancer Genome Atlas (TCGA) were employed to validate results. Lastly, we used cBioPortal software to examine the hub genetic alterations.
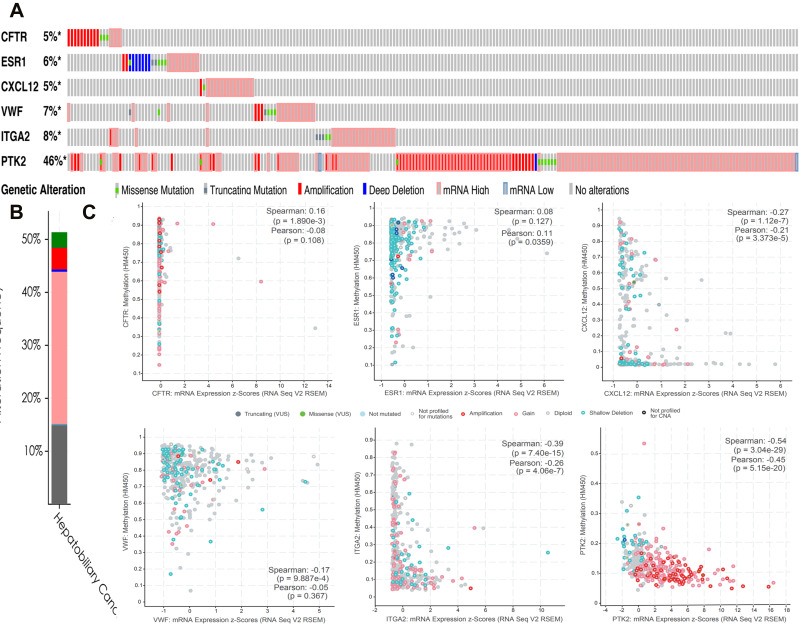
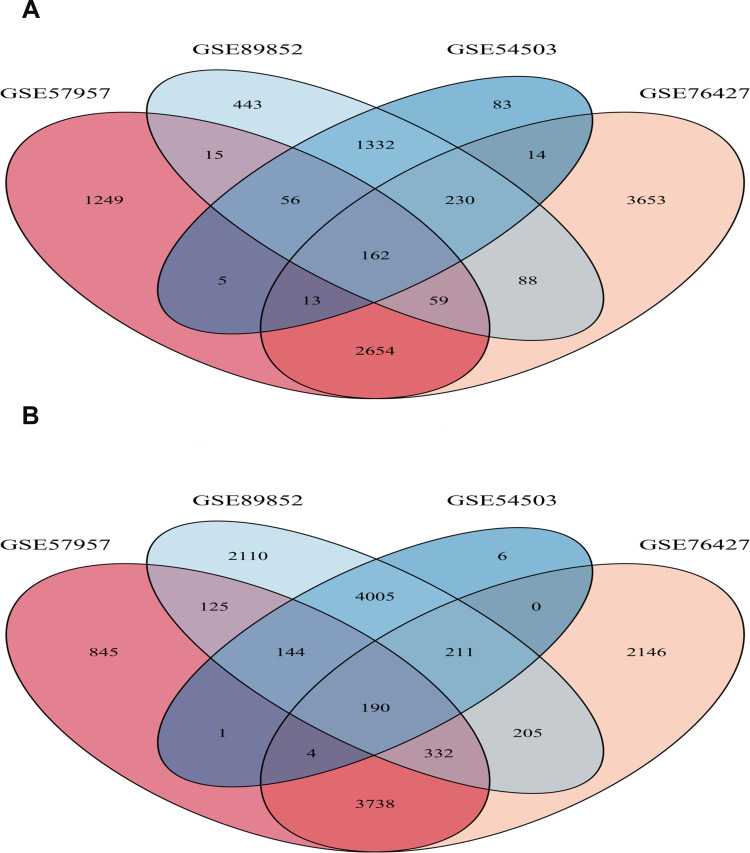
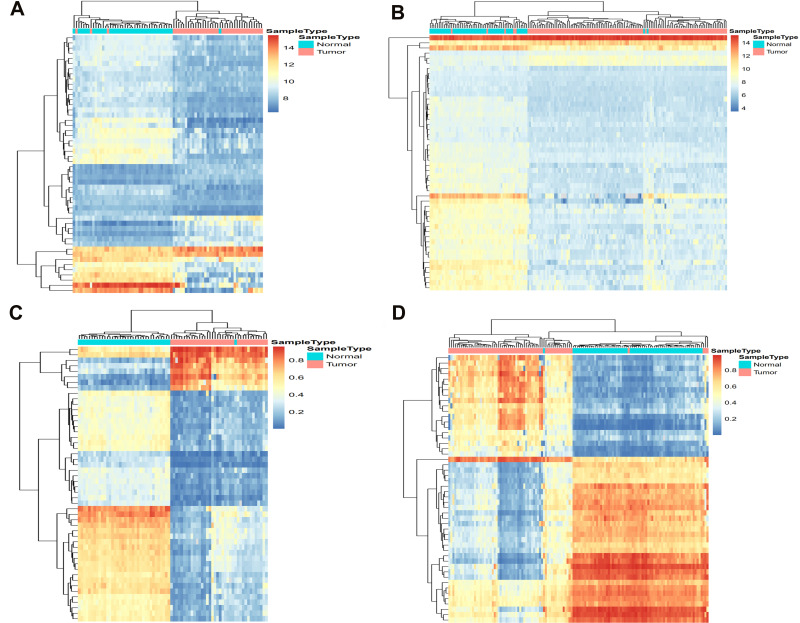
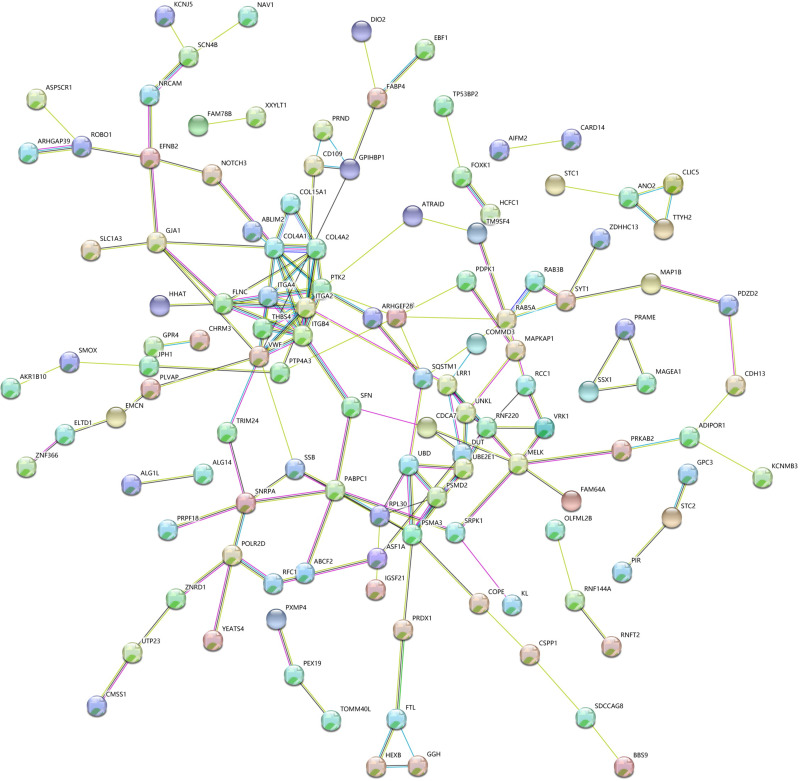
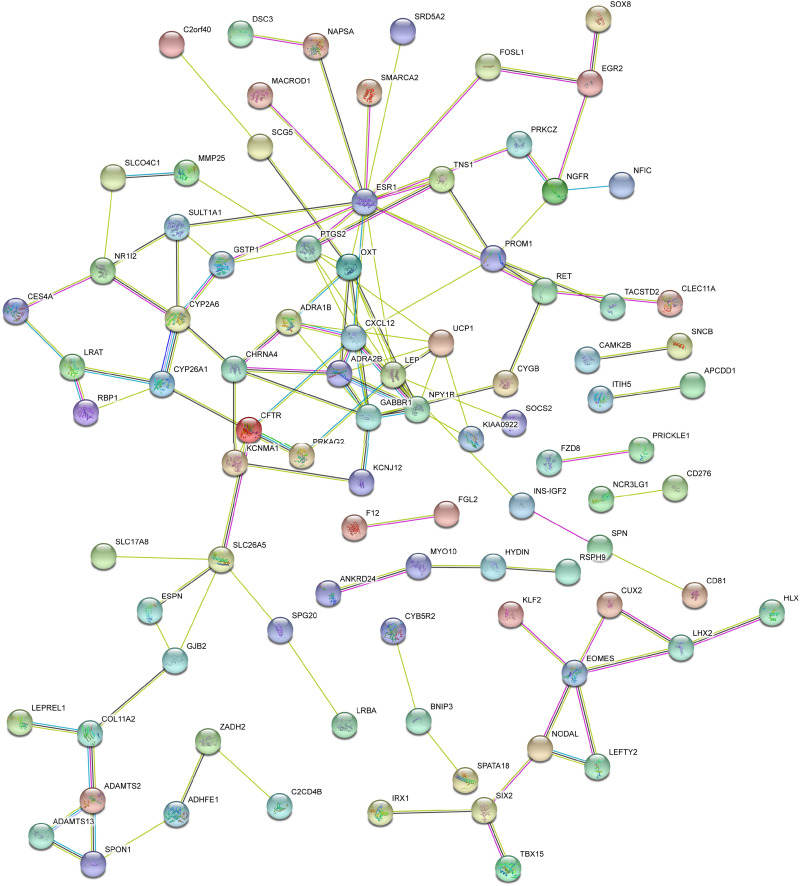
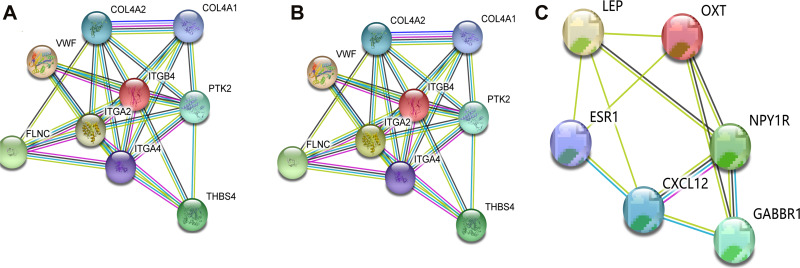
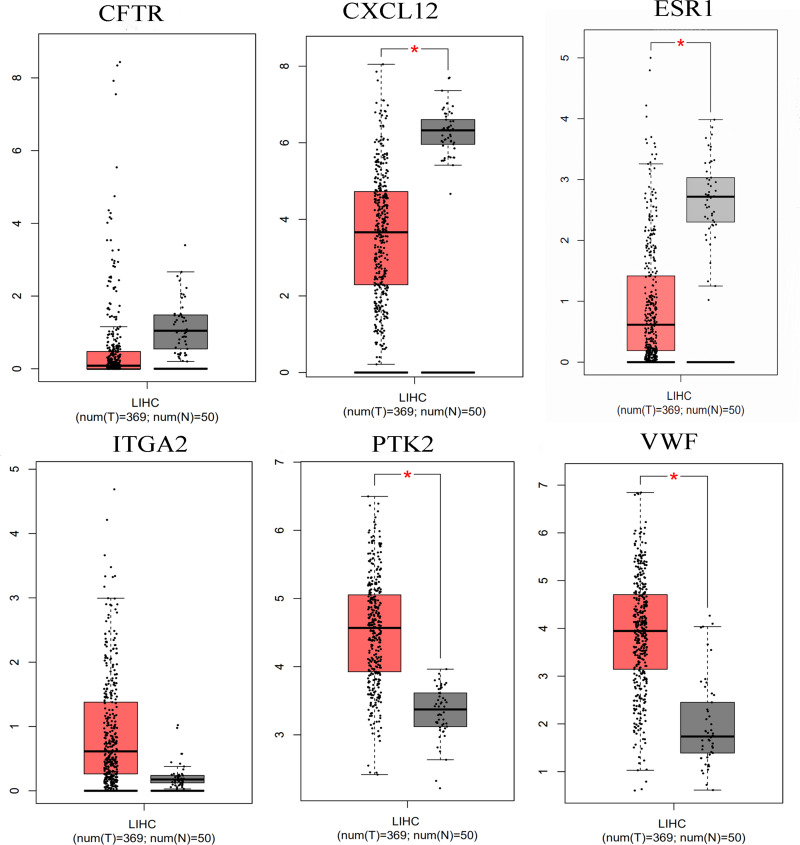
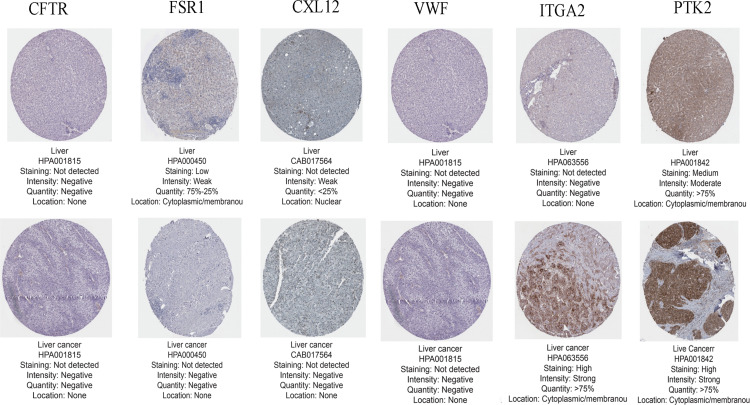
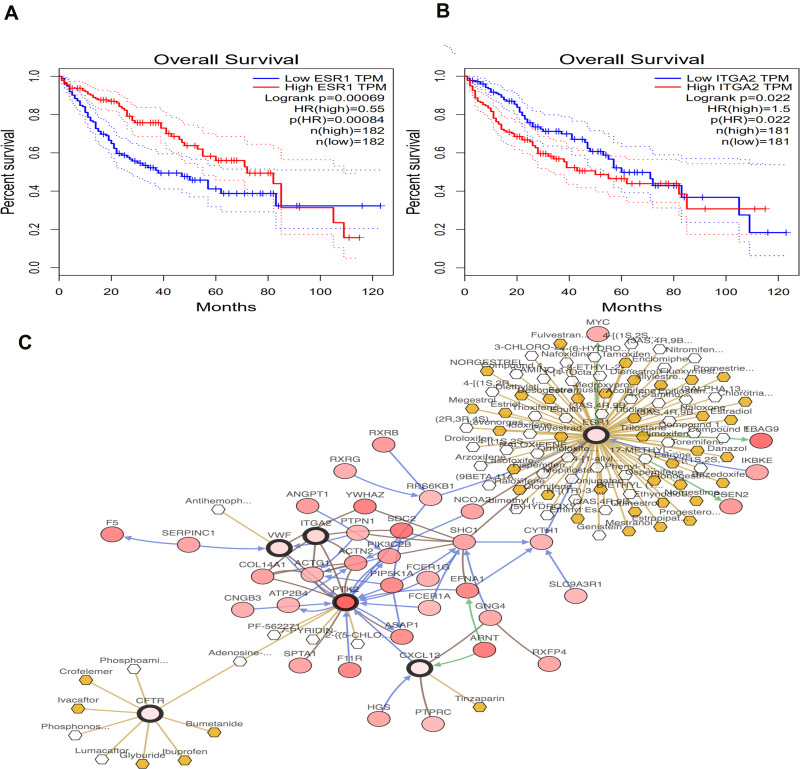
We identified 162 hypermethylated, down-regulated genes and 190 hypomethylated, up-regulated genes. Up-regulated genes with low methylation were enriched in biological processes, such as keratinocyte proliferation, and calcium homeostasis. Pathway analysis was enriched in the AMPK and PI3K-Akt signaling pathways. The PPI network identified PTK2, VWF, and ITGA2 as hypomethylated, high-expressing hub genes. Down-regulated genes with high methylation were related to responses to peptide hormones and estradiol, multi-multicellular organism process. Pathway analysis indicated enrichment in camp, oxytocin signaling pathways. The PPI network identified CFTR, ESR1, and CXCL12 as hypermethylated, low-expressing hub genes. Upon verification in TCGA databases, we found that the expression and methylation statuses of the hub genes changed significantly, and it was consistent with our results.
The novel abnormally MDEGs and pathways in HCC were identified. These results helped us further understand the molecular mechanisms underlying HCC invasion, metastasis, and development. Hub genes can serve as biomarkers for an accurate diagnosis and treatment of HCC, and PTK2, VWF, ITGA2, CFTR, ESR1, and CXCL12 are included.
为了鉴定肝细胞癌(HCC)中异常甲基化的差异表达基因(MDEGs),本研究整合了四个基因芯片数据集以探究肿瘤发生的基本机制。
我们从基因表达综合数据库(GEO)获取了表达谱(GSE76427、GSE57957)和甲基化谱(GSE89852、GSE54503)。使用R软件鉴定异常MDEGs。我们使用clusterProfiler软件包进行功能和通路富集分析。利用String数据库构建蛋白质-蛋白质相互作用(PPI)网络,并在Cytoscape中进行可视化。采用MCODE进行模块分析。此外,利用基因表达谱交互分析(GEPIA)和癌症基因组图谱(TCGA)验证结果。最后,我们使用cBioPortal软件检查核心基因改变。
我们鉴定出162个高甲基化、下调基因和190个低甲基化、上调基因。低甲基化的上调基因在生物学过程中富集,如角质形成细胞增殖和钙稳态。通路分析在AMPK和PI3K-Akt信号通路中富集。PPI网络确定PTK2、VWF和ITGA2为低甲基化、高表达的核心基因。高甲基化的下调基因与对肽激素和雌二醇的反应、多细胞生物体过程有关。通路分析表明在cAMP、催产素信号通路中富集。PPI网络确定CFTR、ESR1和CXCL12为高甲基化、低表达的核心基因。在TCGA数据库中验证后,我们发现核心基因的表达和甲基化状态发生了显著变化,且与我们的结果一致。
鉴定出了HCC中新型的异常MDEGs和通路。这些结果有助于我们进一步了解HCC侵袭、转移和发展的分子机制。核心基因可作为HCC准确诊断和治疗的生物标志物,其中包括PTK2、VWF、ITGA2、CFTR、ESR1和CXCL12。