Föhr Karl Josef, Nastos Ariadni, Fauler Michael, Zimmer Thomas, Jungwirth Bettina, Messerer David Alexander Christian
Department of Anesthesiology and Intensive Care Medicine, University Hospital Ulm, Ulm, Germany.
Department of General Physiology, Ulm University, Ulm, Germany.
Front Pharmacol. 2021 Feb 25;12:622489. doi: 10.3389/fphar.2021.622489. eCollection 2021.
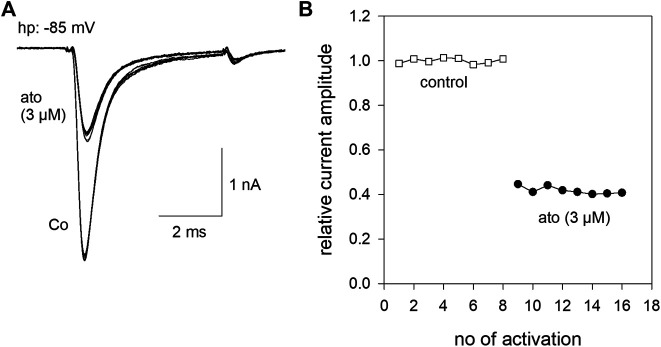
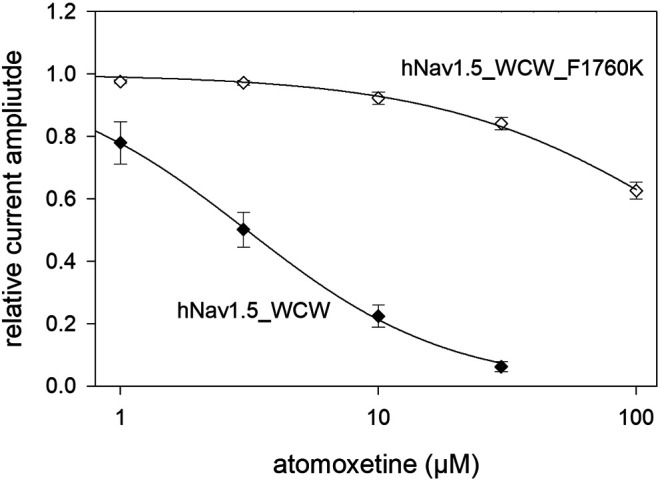
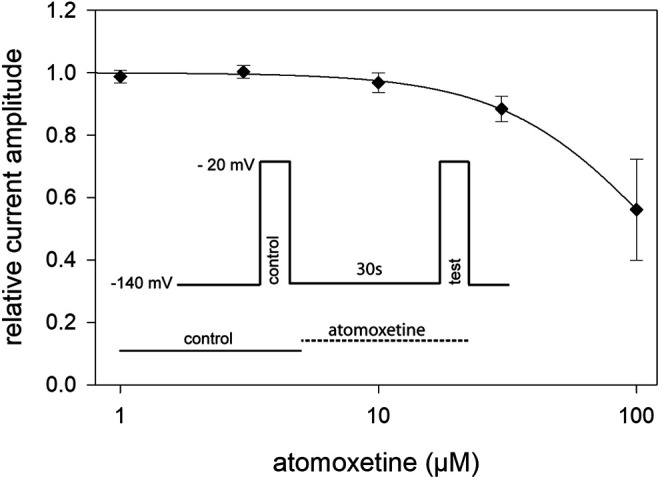
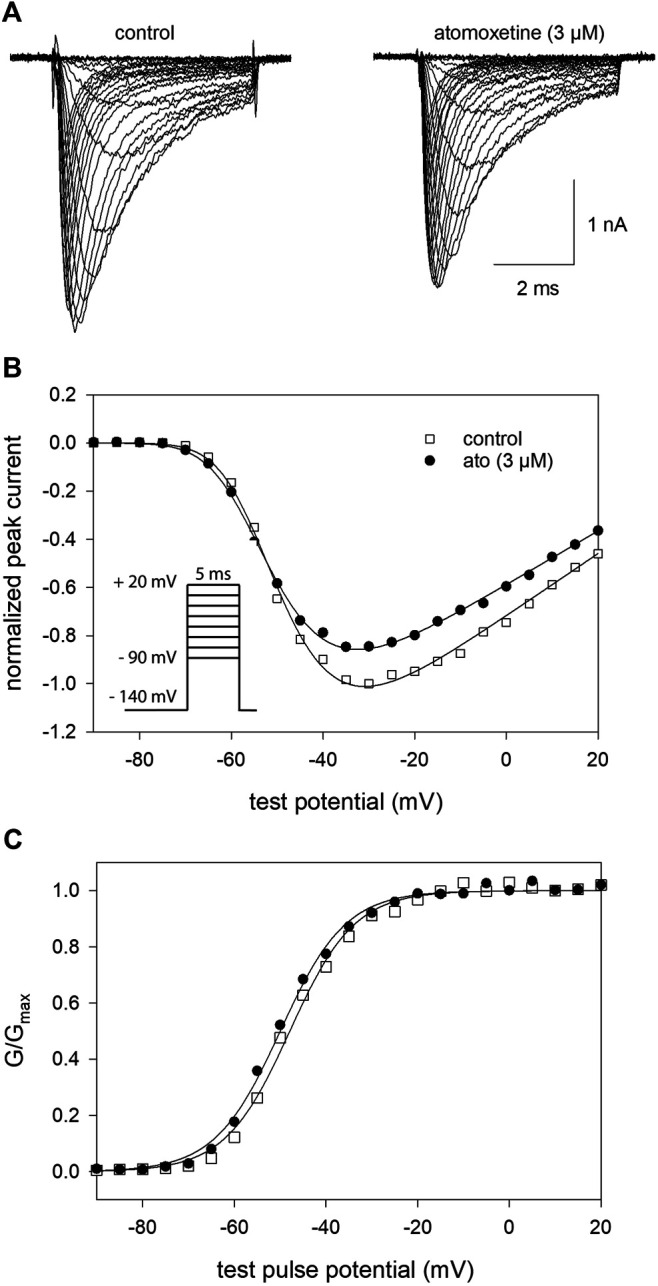
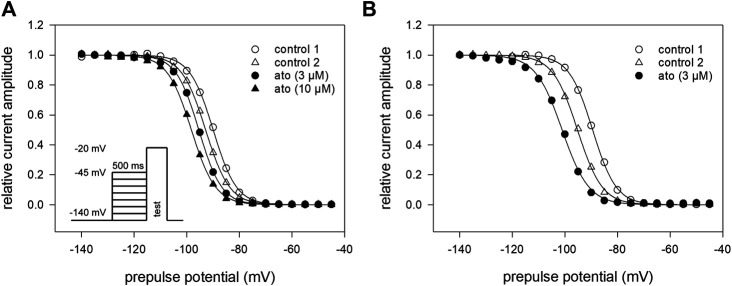
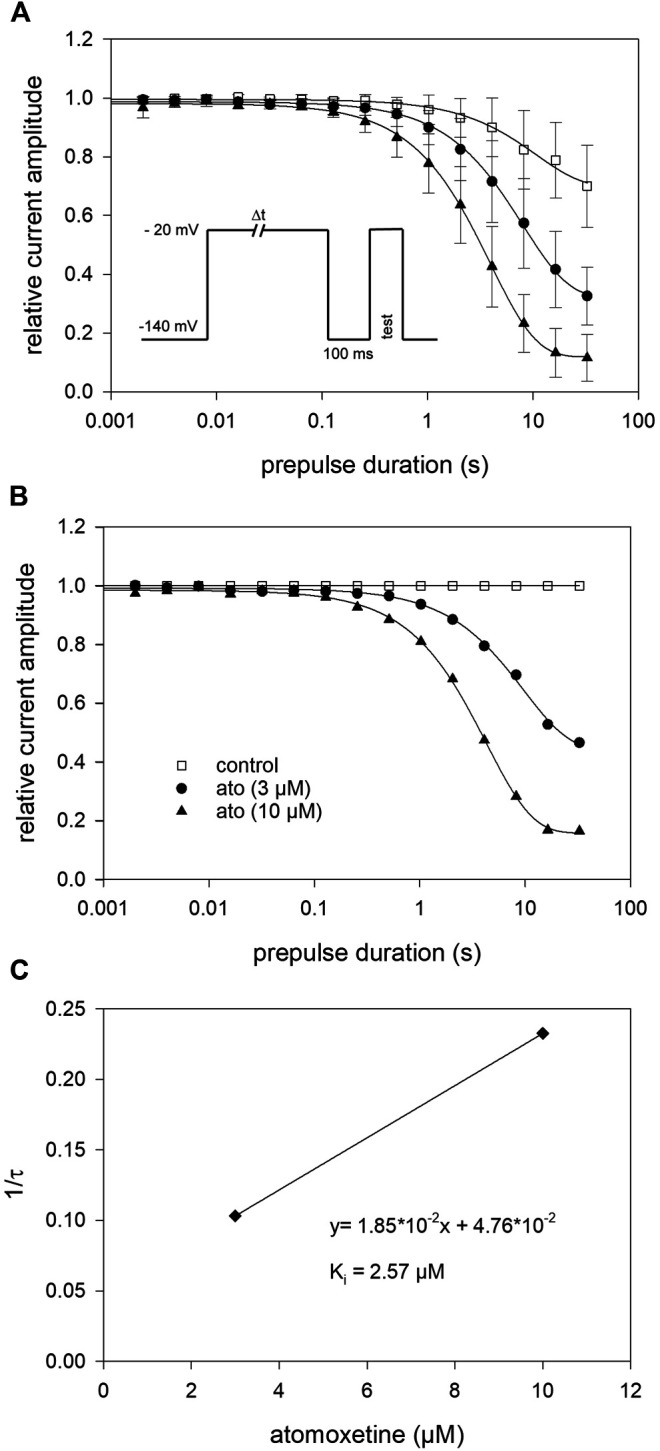
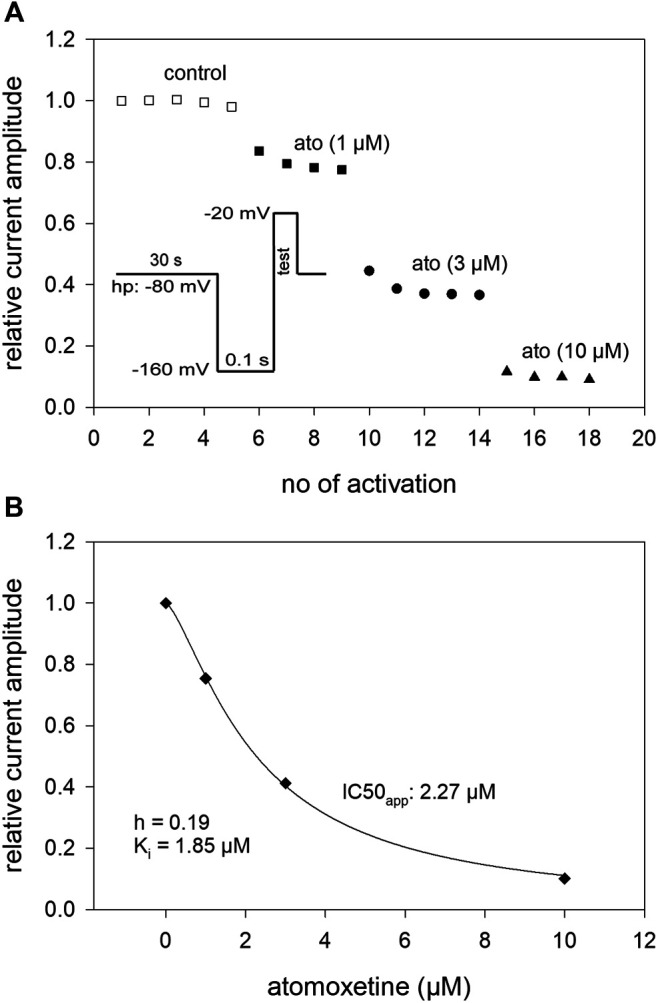
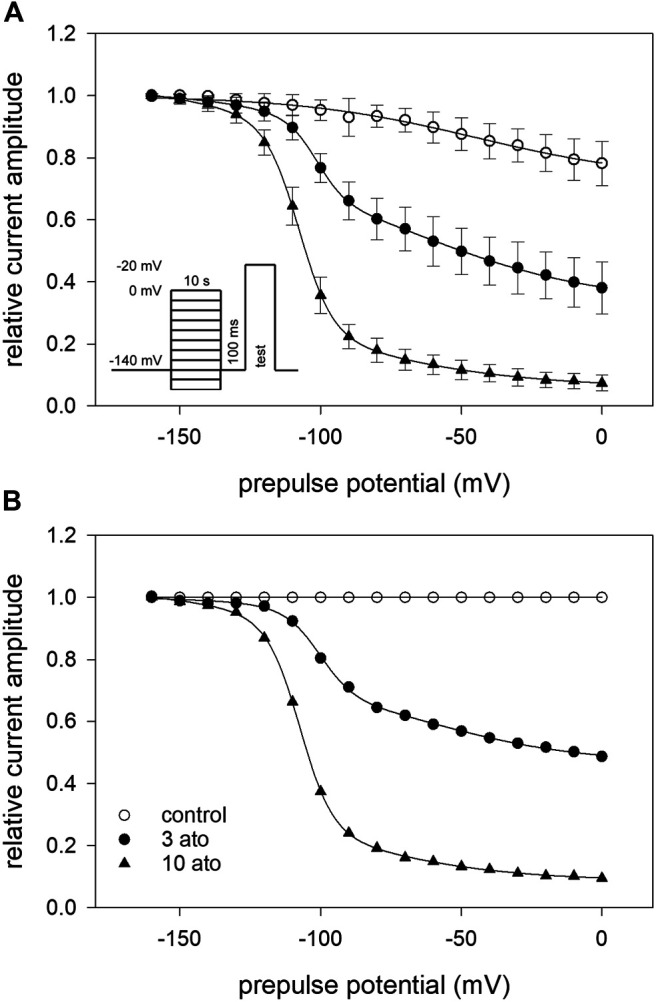
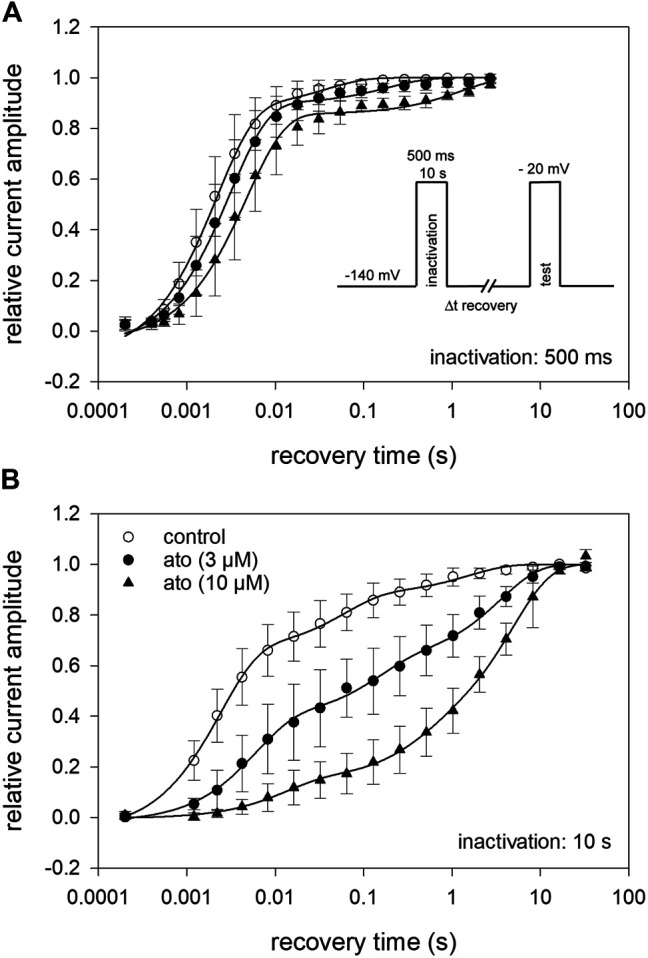
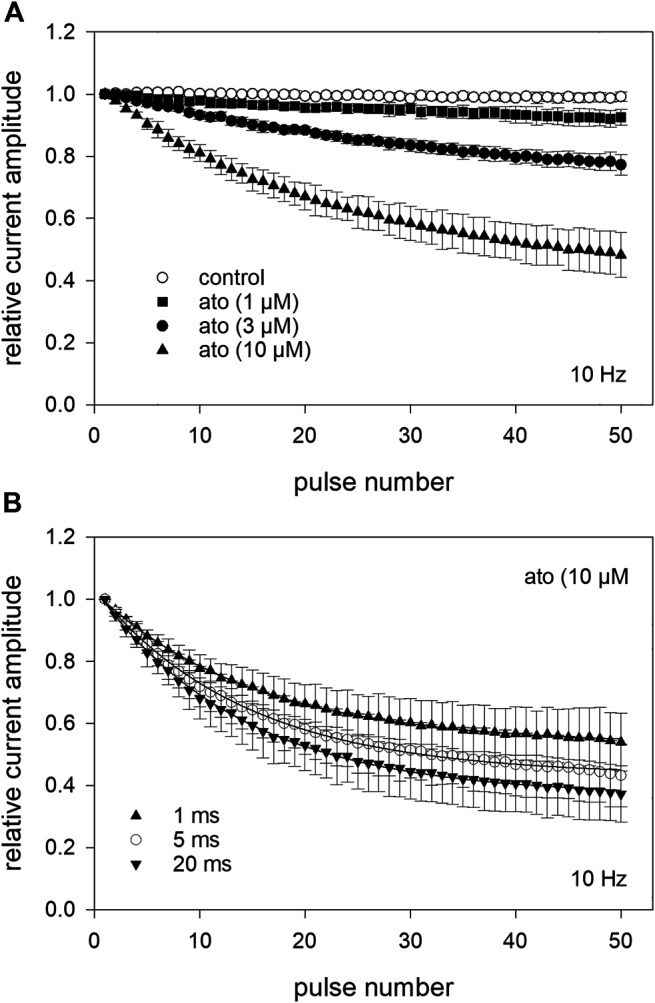
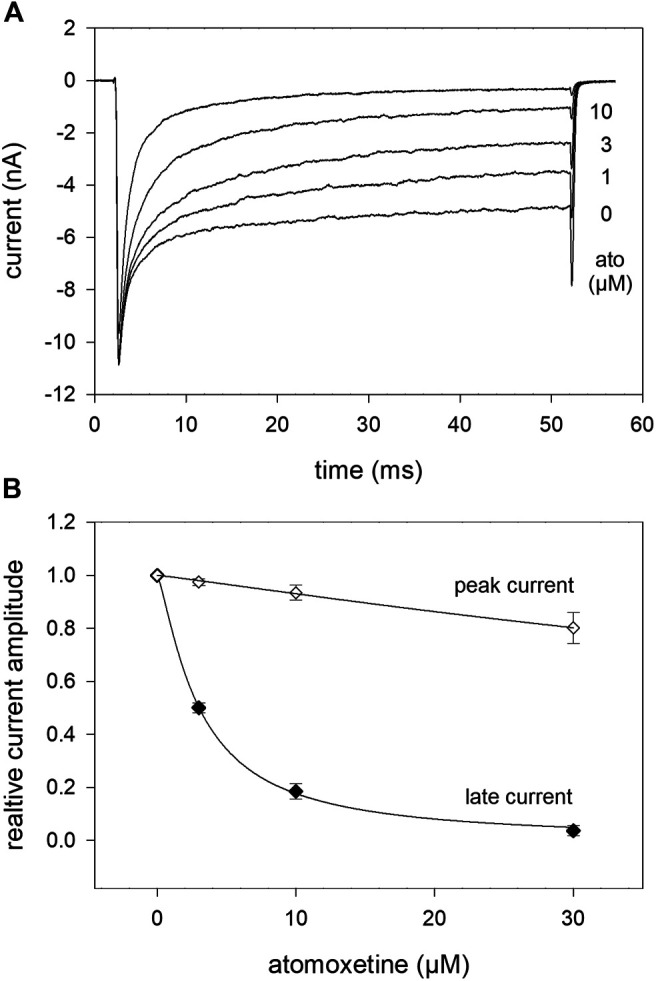
Atomoxetine, a neuroactive drug, is approved for the treatment of attention-deficit/hyperactivity disorder (ADHD). It is primarily known as a high affinity blocker of the noradrenaline transporter, whereby its application leads to an increased level of the corresponding neurotransmitter in different brain regions. However, the concentrations used to obtain clinical effects are much higher than those which are required to block the transporter system. Thus, off-target effects are likely to occur. In this way, we previously identified atomoxetine as blocker of NMDA receptors. As many psychotropic drugs give rise to sudden death of cardiac origin, we now tested the hypothesis whether atomoxetine also interacts with voltage-gated sodium channels of heart muscle type in clinically relevant concentrations. Electrophysiological experiments were performed by means of the patch-clamp technique at human heart muscle sodium channels (hNav1.5) heterogeneously expressed in human embryonic kidney cells. Atomoxetine inhibited sodium channels in a state- and use-dependent manner. Atomoxetine had only a weak affinity for the resting state of the hNav1.5 (Kr: ∼ 120 µM). The efficacy of atomoxetine strongly increased with membrane depolarization, indicating that the inactivated state is an important target. A hallmark of this drug was its slow interaction. By use of different experimental settings, we concluded that the interaction occurs with the slow inactivated state as well as by slow kinetics with the fast-inactivated state. Half-maximal effective concentrations (2-3 µM) were well within the concentration range found in plasma of treated patients. Atomoxetine also interacted with the open channel. However, the interaction was not fast enough to accelerate the time constant of fast inactivation. Nevertheless, when using the inactivation-deficient hNav1.5_I408W_L409C_A410W mutant, we found that the persistent late current was blocked half maximal at about 3 µM atomoxetine. The interaction most probably occurred via the local anesthetic binding site. Atomoxetine inhibited sodium channels at a similar concentration as it is used for the treatment of ADHD. Due to its slow interaction and by inhibiting the late current, it potentially exerts antiarrhythmic properties.
托莫西汀是一种神经活性药物,已被批准用于治疗注意力缺陷多动障碍(ADHD)。它主要作为去甲肾上腺素转运体的高亲和力阻滞剂而为人所知,其应用会导致不同脑区中相应神经递质水平升高。然而,用于获得临床效果的浓度远高于阻断转运体系统所需的浓度。因此,可能会出现脱靶效应。通过这种方式,我们之前确定托莫西汀是NMDA受体的阻滞剂。由于许多精神药物会导致心源性猝死,我们现在测试了一个假设,即托莫西汀在临床相关浓度下是否也与心肌型电压门控钠通道相互作用。通过膜片钳技术在人胚胎肾细胞中异源表达的人心脏肌肉钠通道(hNav1.5)上进行电生理实验。托莫西汀以状态和使用依赖性方式抑制钠通道。托莫西汀对hNav1.5的静息状态亲和力较弱(解离常数:约120μM)。托莫西汀的效力随着膜去极化而强烈增加,表明失活状态是一个重要靶点。这种药物的一个特点是其相互作用缓慢。通过使用不同的实验设置,我们得出结论,这种相互作用发生在缓慢失活状态以及与快速失活状态的缓慢动力学过程中。半数最大有效浓度(2 - 3μM)完全在治疗患者血浆中发现的浓度范围内。托莫西汀也与开放通道相互作用。然而,这种相互作用不够快,无法加速快速失活的时间常数。尽管如此,当使用失活缺陷型hNav1.5_I408W_L409C_A410W突变体时,我们发现约3μM托莫西汀可使持续性晚电流半数最大阻断。这种相互作用很可能通过局部麻醉药结合位点发生。托莫西汀在与治疗ADHD相同的浓度下抑制钠通道。由于其相互作用缓慢并抑制晚电流,它可能具有抗心律失常特性。