University of Maine, Orono, Maine, United States of America.
PLoS Comput Biol. 2021 Mar 18;17(3):e1008674. doi: 10.1371/journal.pcbi.1008674. eCollection 2021 Mar.
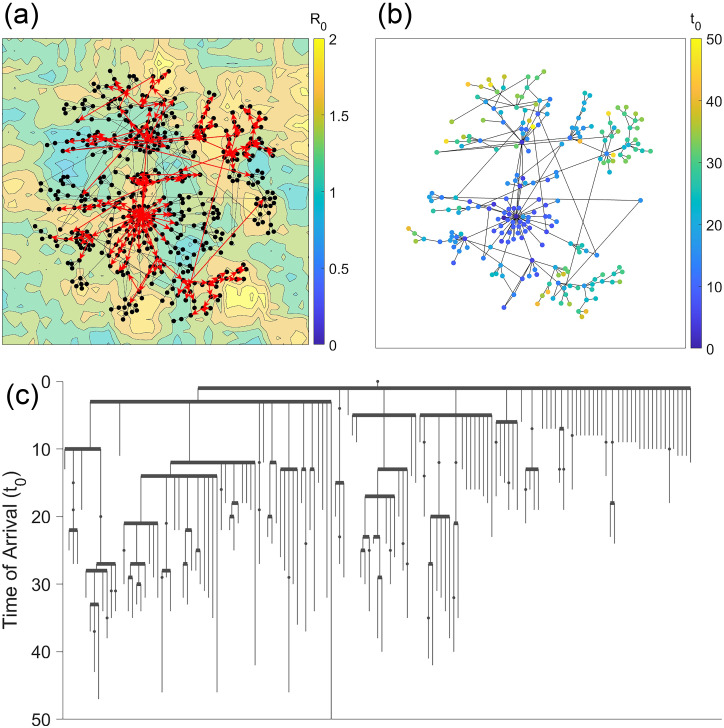
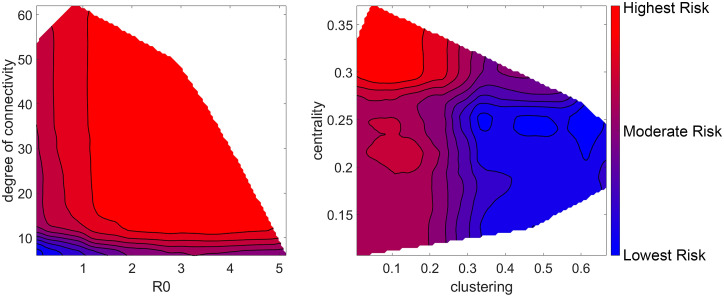
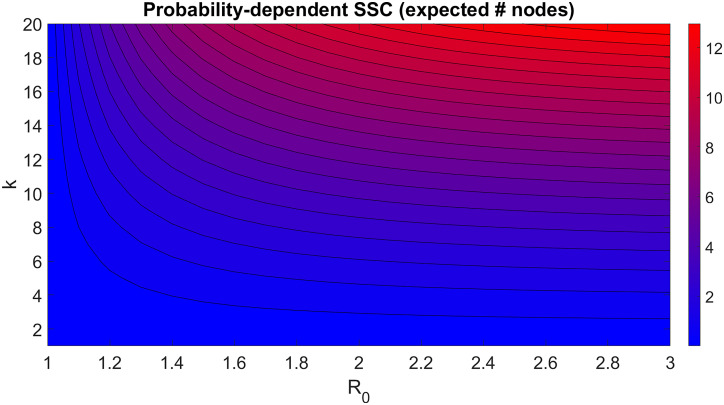
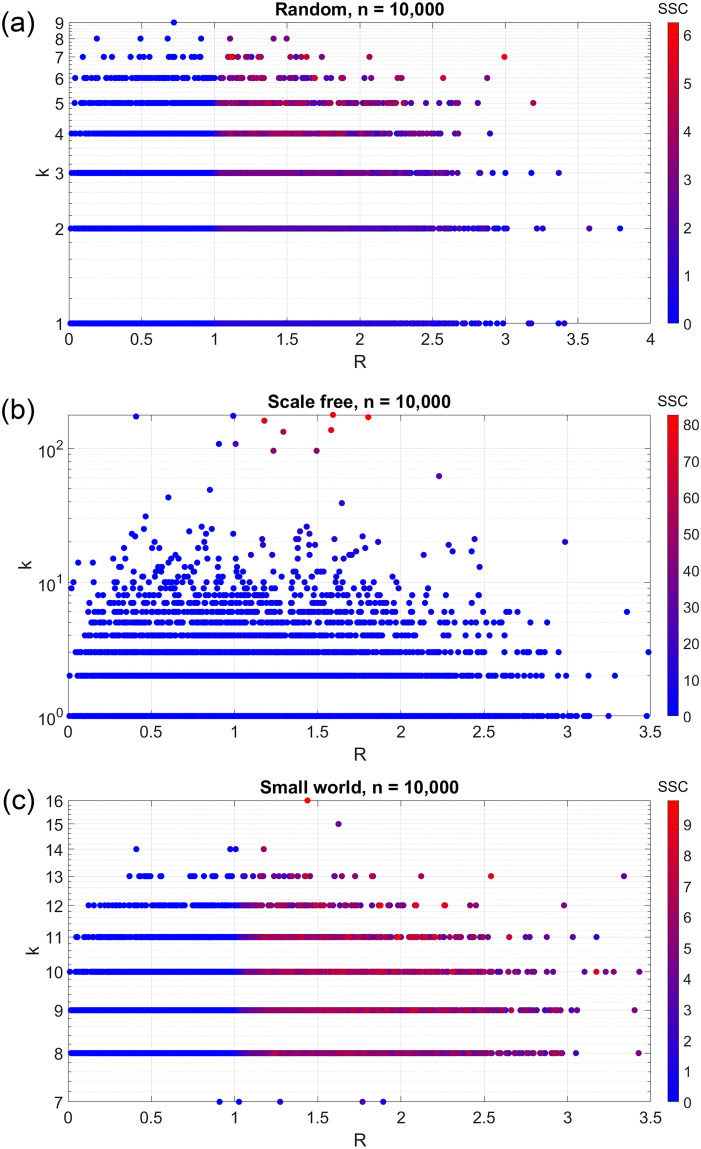
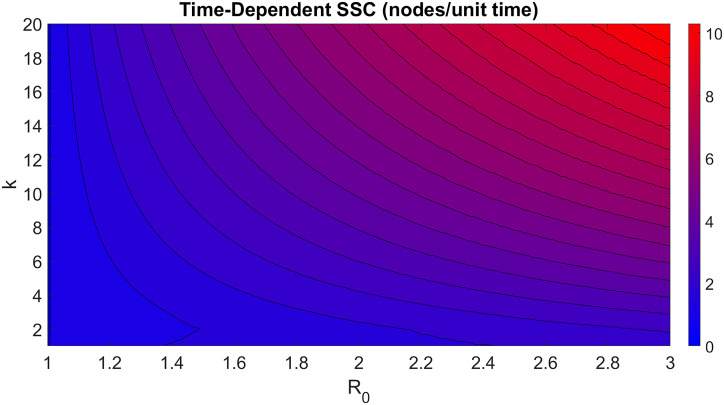
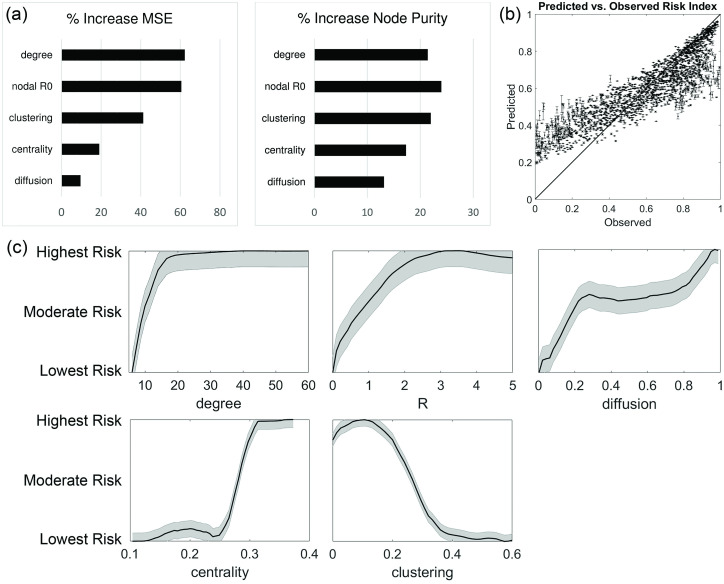
Disease epidemic outbreaks on human metapopulation networks are often driven by a small number of superspreader nodes, which are primarily responsible for spreading the disease throughout the network. Superspreader nodes typically are characterized either by their locations within the network, by their degree of connectivity and centrality, or by their habitat suitability for the disease, described by their reproduction number (R). Here we introduce a model that considers simultaneously the effects of network properties and R on superspreaders, as opposed to previous research which considered each factor separately. This type of model is applicable to diseases for which habitat suitability varies by climate or land cover, and for direct transmitted diseases for which population density and mitigation practices influences R. We present analytical models that quantify the superspreader capacity of a population node by two measures: probability-dependent superspreader capacity, the expected number of neighboring nodes to which the node in consideration will randomly spread the disease per epidemic generation, and time-dependent superspreader capacity, the rate at which the node spreads the disease to each of its neighbors. We validate our analytical models with a Monte Carlo analysis of repeated stochastic Susceptible-Infected-Recovered (SIR) simulations on randomly generated human population networks, and we use a random forest statistical model to relate superspreader risk to connectivity, R, centrality, clustering, and diffusion. We demonstrate that either degree of connectivity or R above a certain threshold are sufficient conditions for a node to have a moderate superspreader risk factor, but both are necessary for a node to have a high-risk factor. The statistical model presented in this article can be used to predict the location of superspreader events in future epidemics, and to predict the effectiveness of mitigation strategies that seek to reduce the value of R, alter host movements, or both.
人类集合种群网络上的疾病疫情爆发通常是由少数超级传播者节点驱动的,这些节点主要负责在网络中传播疾病。超级传播者节点通常具有以下特征:在网络中的位置、连接度和中心度,或者其对疾病的栖息地适宜性,由其繁殖数(R)描述。在这里,我们引入了一个模型,该模型同时考虑了网络属性和 R 对超级传播者的影响,而不是之前的研究分别考虑每个因素。这种模型适用于栖息地适宜性随气候或土地覆盖变化的疾病,以及人口密度和缓解措施影响 R 的直接传播疾病。我们提出了两个衡量标准来量化人口节点的超级传播者能力的分析模型:概率相关的超级传播者能力,考虑节点在每次疫情传播中随机传播疾病的预期相邻节点数量,以及时间相关的超级传播者能力,即节点传播疾病到每个邻居的速度。我们通过对随机生成的人类人口网络上重复的随机易感染-感染-恢复(SIR)模拟进行蒙特卡罗分析,验证了我们的分析模型,并使用随机森林统计模型将超级传播者风险与连接度、R、中心度、聚类和扩散联系起来。我们表明,连接度或 R 超过一定阈值是节点具有中等超级传播者风险因素的充分条件,但两者都是节点具有高风险因素的必要条件。本文提出的统计模型可用于预测未来疫情中超级传播者事件的位置,并预测旨在降低 R 值、改变宿主运动或两者兼而有之的缓解策略的有效性。