Du Jian-Kui, Yu Qing, Liu Yu-Jian, Du Shu-Fang, Huang Li-Yang, Xu Dan-Hong, Ni Xin, Zhu Xiao-Yan
National Clinical Research Center for Geriatric Disorders and National International Joint Research Center for Medical Metabolomics, Xiangya Hospital, Central South University, Changsha, Hunan, China.
Department of Physiology, Navy Medical University, Shanghai, China.
Theranostics. 2021 Feb 20;11(9):4207-4231. doi: 10.7150/thno.48530. eCollection 2021.
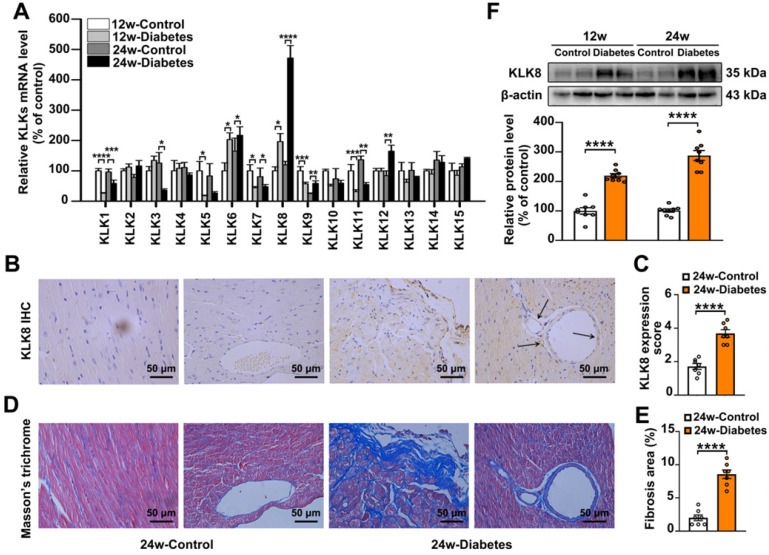
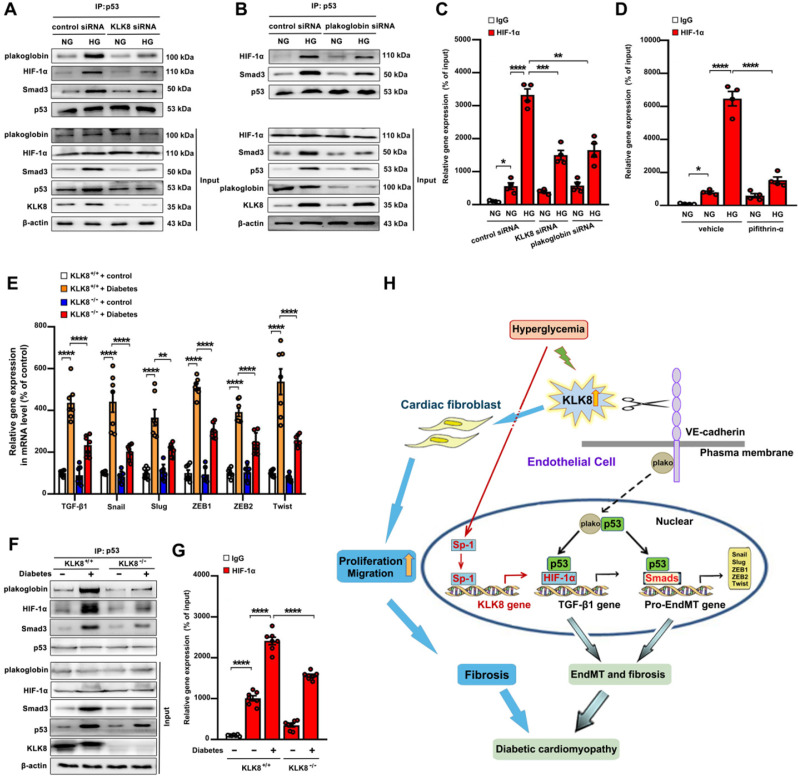
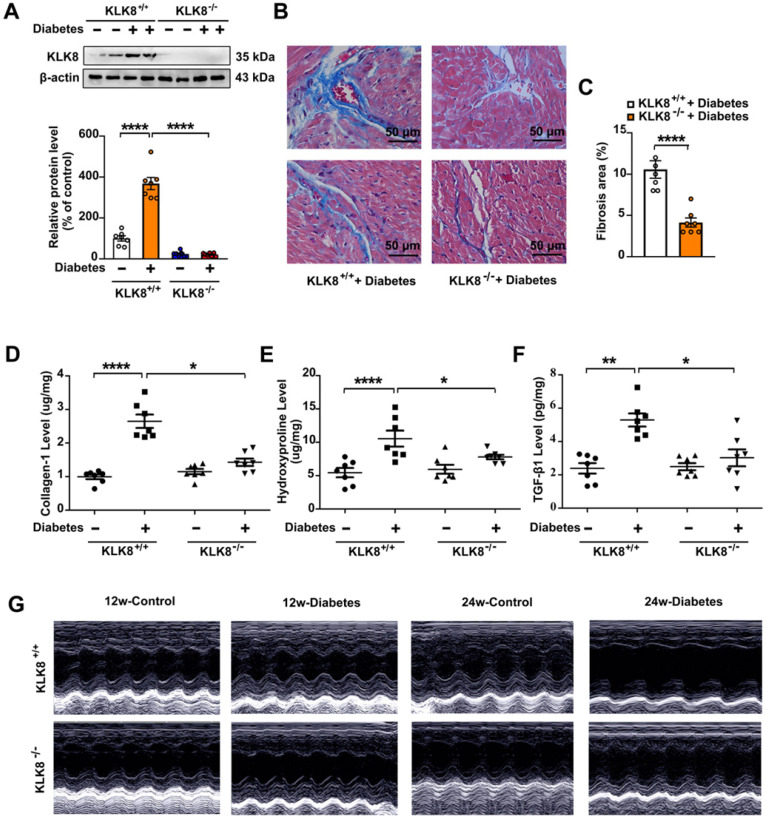
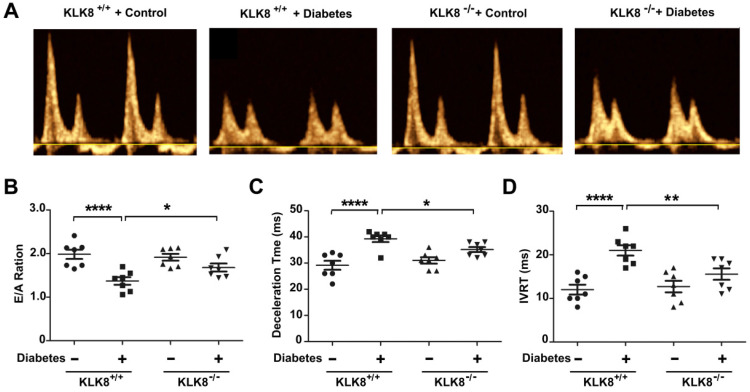
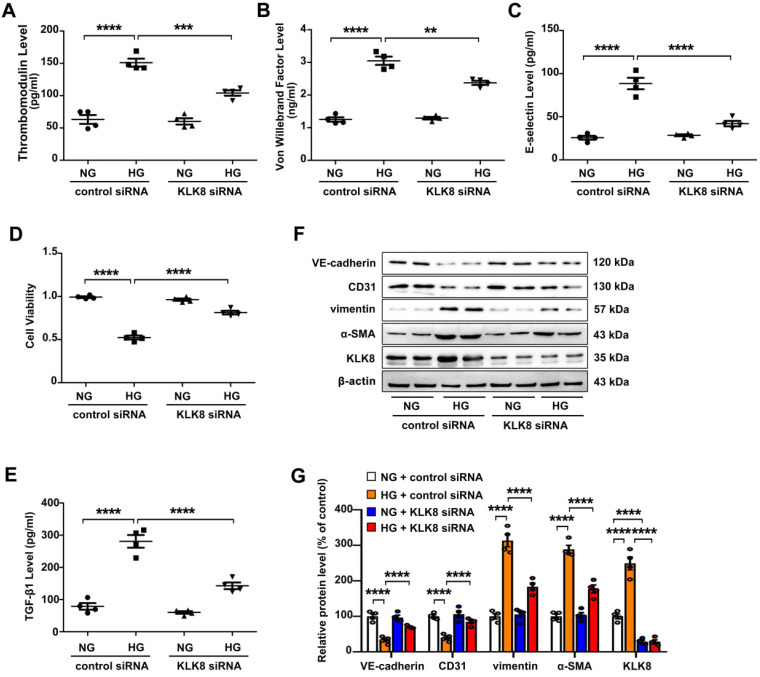
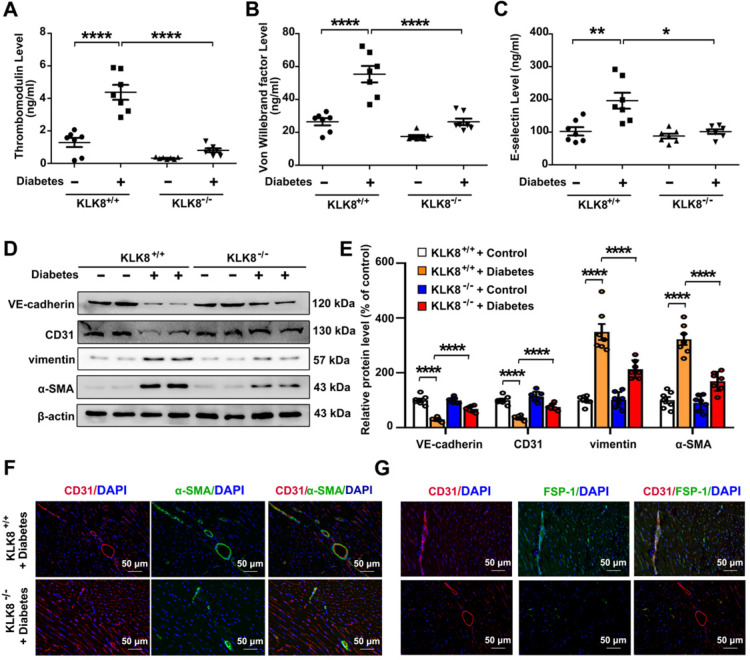
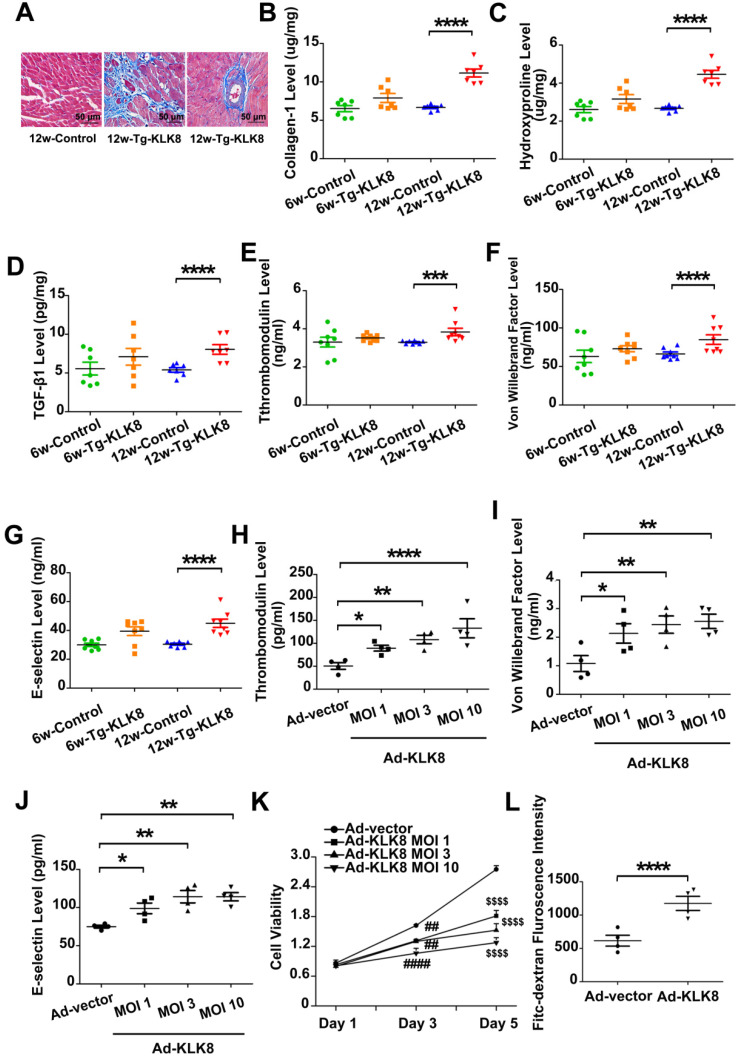
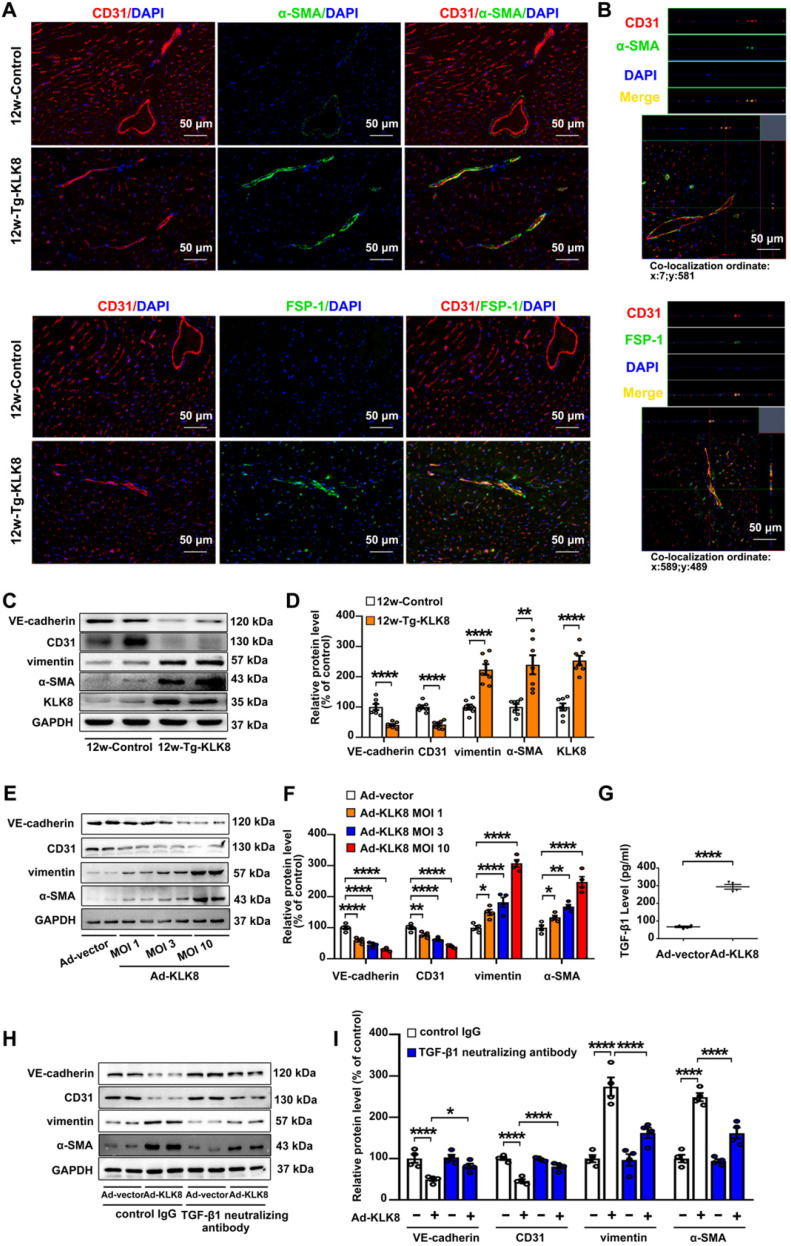
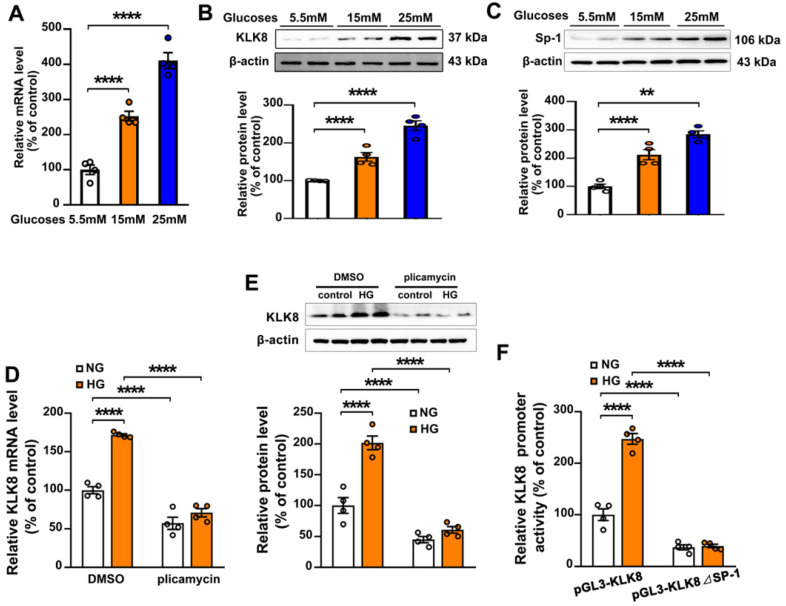
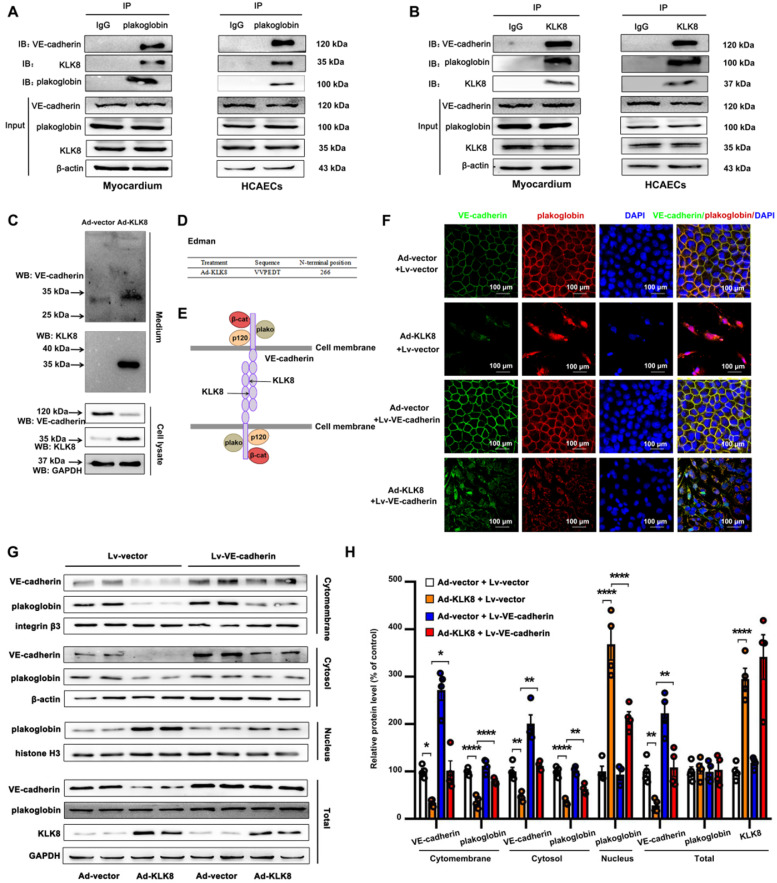
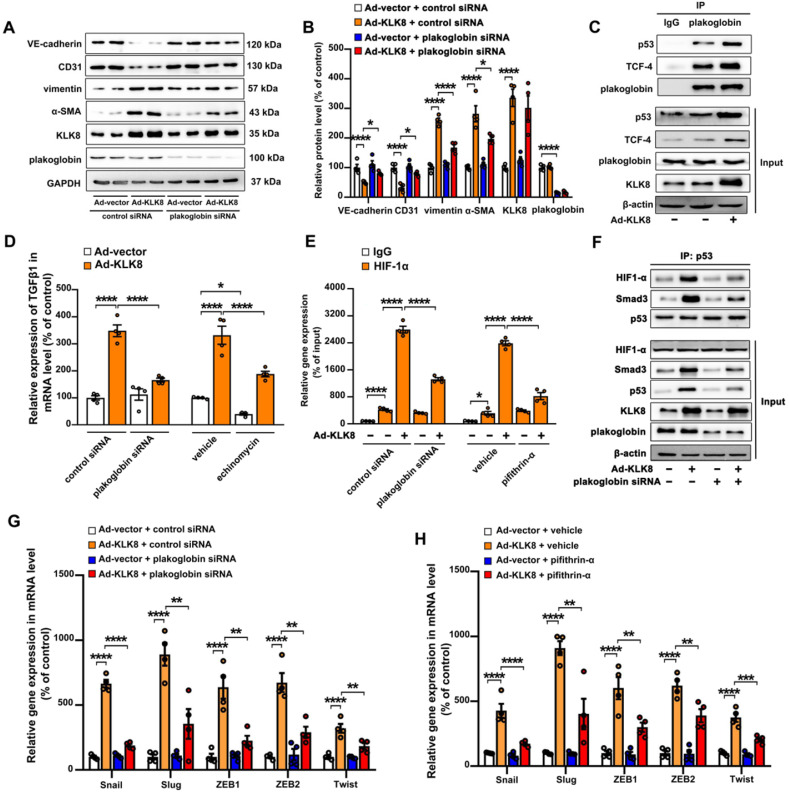
Among all the diabetic complications, diabetic cardiomyopathy, which is characterized by myocyte loss and myocardial fibrosis, is the leading cause of mortality and morbidity in diabetic patients. Tissue kallikrein-related peptidases (KLKs) are secreted serine proteases, that have distinct and overlapping roles in the pathogenesis of cardiovascular diseases. However, whether KLKs are involved in the development of diabetic cardiomyopathy remains unknown.The present study aimed to determine the role of a specific KLK in the initiation of endothelial-to-mesenchymal transition (EndMT) during the pathogenesis of diabetic cardiomyopathy. By screening gene expression profiles of KLKs, it was found that KLK8 was highly induced in the myocardium of mice with streptozotocin-induced diabetes. KLK8 deficiency attenuated diabetic cardiac fibrosis, and rescued the impaired cardiac function in diabetic mice. Small interfering RNA (siRNA)-mediated KLK8 knockdown significantly attenuated high glucose-induced endothelial damage and EndMT in human coronary artery endothelial cells (HCAECs). Diabetes-induced endothelial injury and cardiac EndMT were significantly alleviated in KLK8-deficient mice. In addition, transgenic overexpression of KLK8 led to interstitial and perivascular cardiac fibrosis, endothelial injury and EndMT in the heart. Adenovirus-mediated overexpression of KLK8 (Ad-KLK8) resulted in increases in endothelial cell damage, permeability and transforming growth factor (TGF)-β1 release in HCAECs. KLK8 overexpression also induced EndMT in HCAECs, which was alleviated by a TGF-β1-neutralizing antibody. A specificity protein-1 (Sp-1) consensus site was identified in the human KLK8 promoter and was found to mediate the high glucose-induced KLK8 expression. Mechanistically, it was identified that the vascular endothelial (VE)-cadherin/plakoglobin complex may associate with KLK8 in HCAECs. KLK8 cleaved the VE-cadherin extracellular domain, thus promoting plakoglobin nuclear translocation. Plakoglobin was required for KLK8-induced EndMT by cooperating with p53. KLK8 overexpression led to plakoglobin-dependent association of p53 with hypoxia inducible factor (HIF)-1α, which further enhanced the transactivation effect of HIF-1α on the TGF-β1 promoter. KLK8 also induced the binding of p53 with Smad3, subsequently promoting pro-EndMT reprogramming via the TGF-β1/Smad signaling pathway in HCAECs. The and findings further demonstrated that high glucose may promote plakoglobin-dependent cooperation of p53 with HIF-1α and Smad3, subsequently increasing the expression of TGF-β1 and the pro-EndMT target genes of the TGF-β1/Smad signaling pathway in a KLK8-dependent manner. The present findings uncovered a novel pro-EndMT mechanism during the pathogenesis of diabetic cardiac fibrosis via the upregulation of KLK8, and may contribute to the development of future KLK8-based therapeutic strategies for diabetic cardiomyopathy.
在所有糖尿病并发症中,以心肌细胞丢失和心肌纤维化为特征的糖尿病性心肌病是糖尿病患者死亡和发病的主要原因。组织激肽释放酶相关肽酶(KLKs)是分泌型丝氨酸蛋白酶,在心血管疾病的发病机制中具有不同但相互重叠的作用。然而,KLKs是否参与糖尿病性心肌病的发生尚不清楚。本研究旨在确定一种特定的KLK在糖尿病性心肌病发病机制中内皮-间充质转化(EndMT)起始过程中的作用。通过筛选KLKs的基因表达谱,发现链脲佐菌素诱导的糖尿病小鼠心肌中KLK8被高度诱导。KLK8缺陷减轻了糖尿病心脏纤维化,并挽救了糖尿病小鼠受损的心脏功能。小干扰RNA(siRNA)介导的KLK8敲低显著减轻了高糖诱导的人冠状动脉内皮细胞(HCAECs)的内皮损伤和EndMT。在KLK8缺陷小鼠中,糖尿病诱导的内皮损伤和心脏EndMT显著减轻。此外,KLK8的转基因过表达导致心脏间质和血管周围纤维化、内皮损伤和EndMT。腺病毒介导的KLK8过表达(Ad-KLK8)导致HCAECs中内皮细胞损伤、通透性增加和转化生长因子(TGF)-β1释放增加。KLK8过表达还诱导了HCAECs中的EndMT,这被TGF-β1中和抗体所减轻。在人KLK8启动子中鉴定出一个特异性蛋白-1(Sp-1)共有位点,发现其介导高糖诱导的KLK8表达。机制上,确定血管内皮(VE)-钙黏蛋白/桥粒斑珠蛋白复合物可能在HCAECs中与KLK8结合。KLK8切割VE-钙黏蛋白的细胞外结构域,从而促进桥粒斑珠蛋白的核转位。桥粒斑珠蛋白通过与p53协同作用参与KLK8诱导的EndMT。KLK8过表达导致桥粒斑珠蛋白依赖的p53与缺氧诱导因子(HIF)-1α的结合,这进一步增强了HIF-1α对TGF-β1启动子的反式激活作用。KLK8还诱导p53与Smad3的结合,随后通过TGF-β1/Smad信号通路促进HCAECs中的促EndMT重编程。这些发现进一步证明,高糖可能促进p53与HIF-1α和Smad3的桥粒斑珠蛋白依赖的协同作用,随后以KLK8依赖的方式增加TGF-β1的表达以及TGF-β1/Smad信号通路的促EndMT靶基因的表达。本研究结果揭示了糖尿病心脏纤维化发病机制中通过上调KLK8产生的一种新的促EndMT机制,并可能有助于未来基于KLK8的糖尿病性心肌病治疗策略的开发。