Department of Molecular Life Sciences, University of Zurich, Zurich, Switzerland.
SIB Swiss Institute of Bioinformatics, University of Zurich, Zurich, Switzerland.
Life Sci Alliance. 2021 Mar 23;4(6). doi: 10.26508/lsa.202001004. Print 2021 Jun.
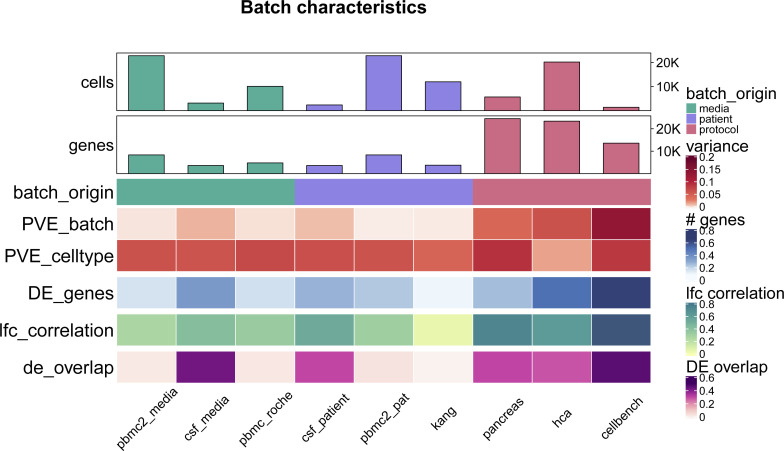
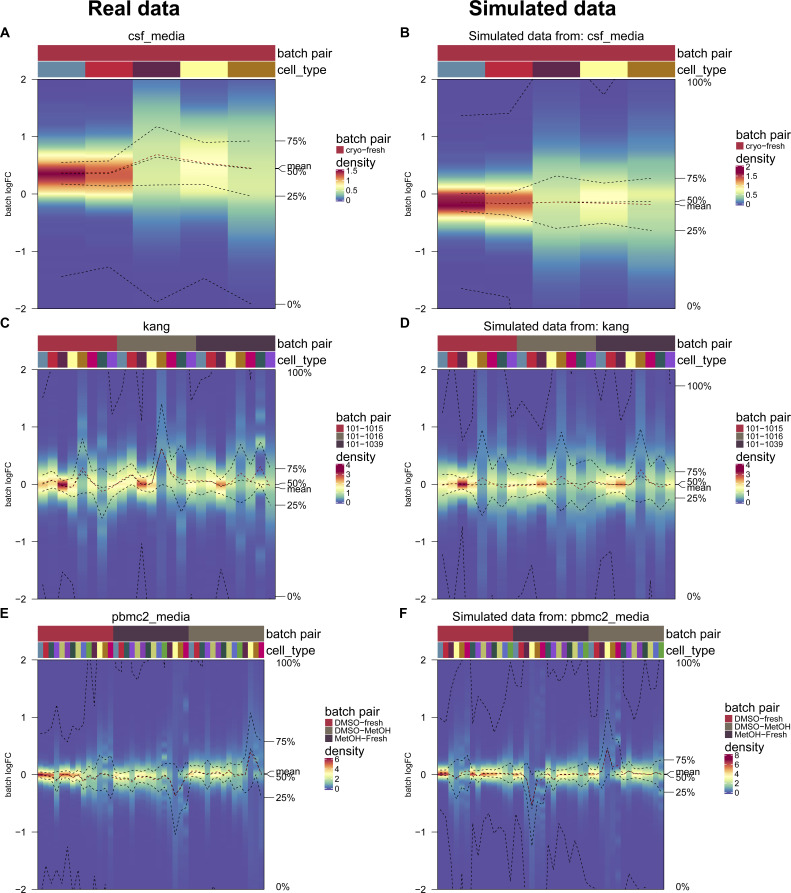
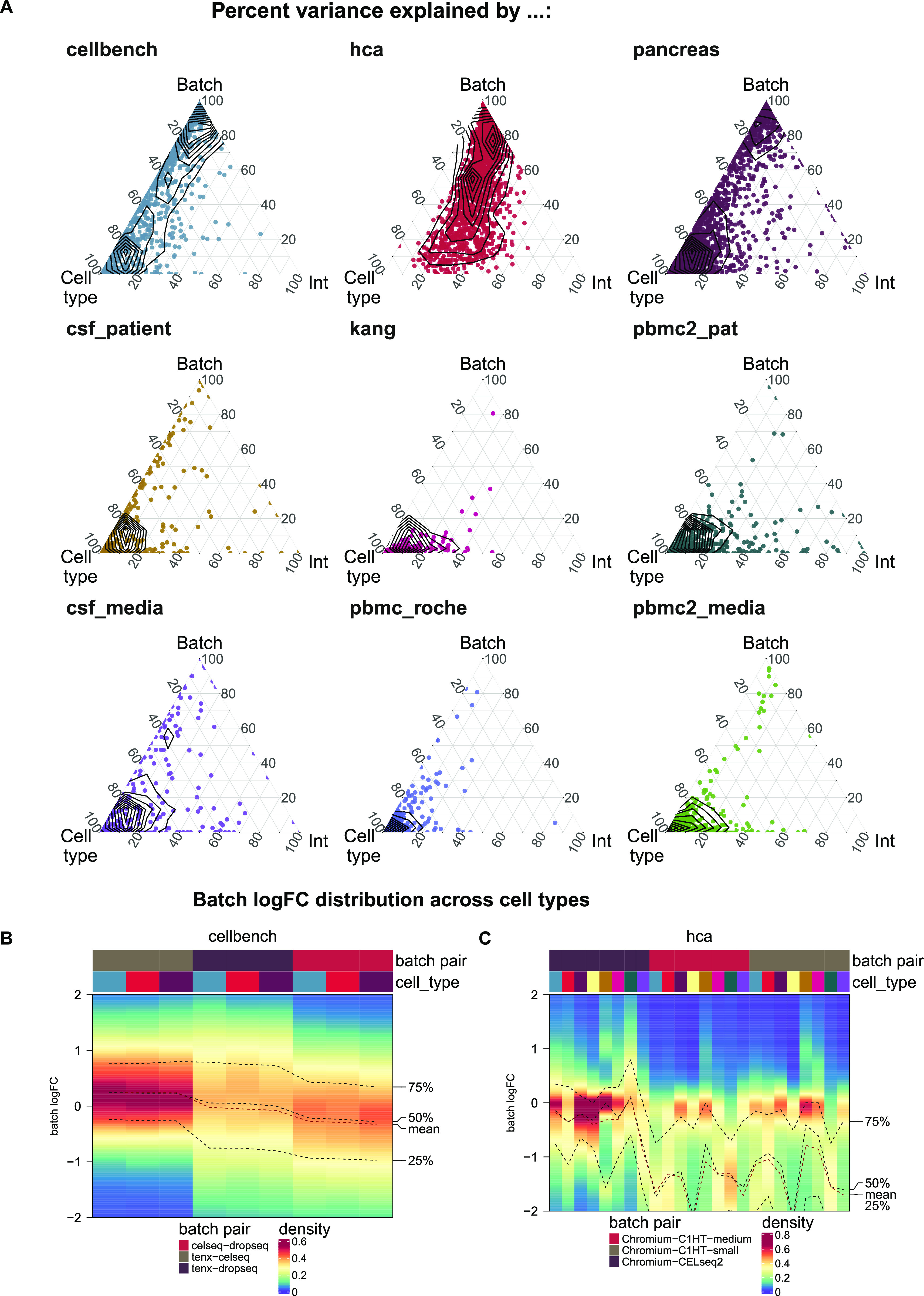
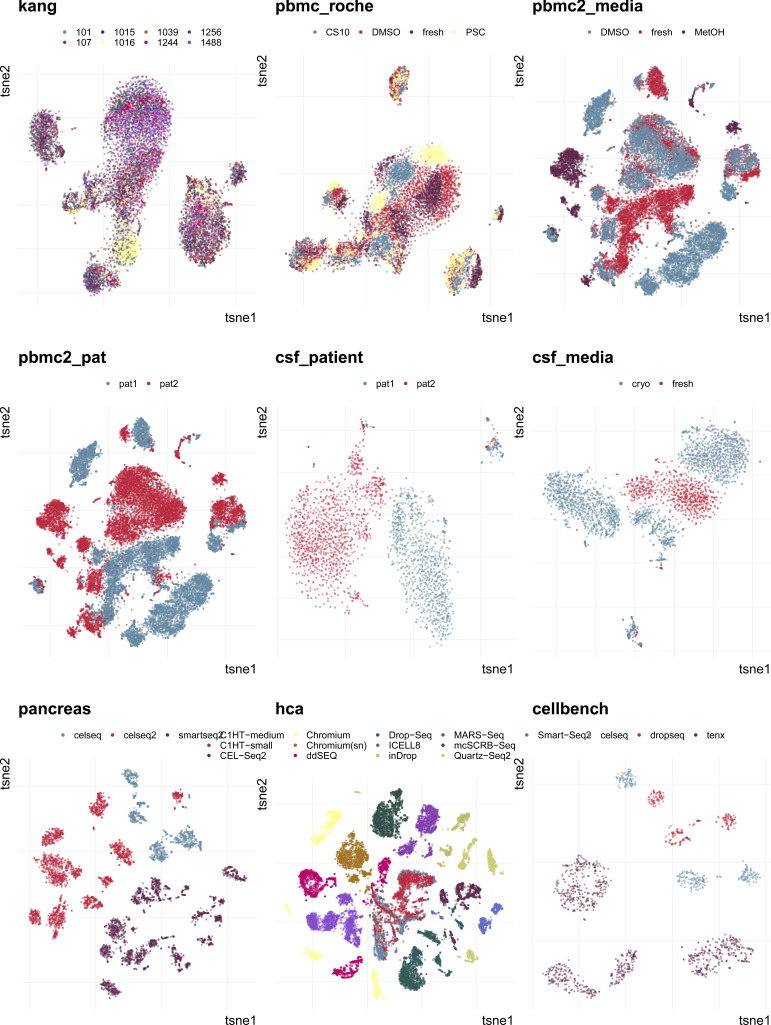
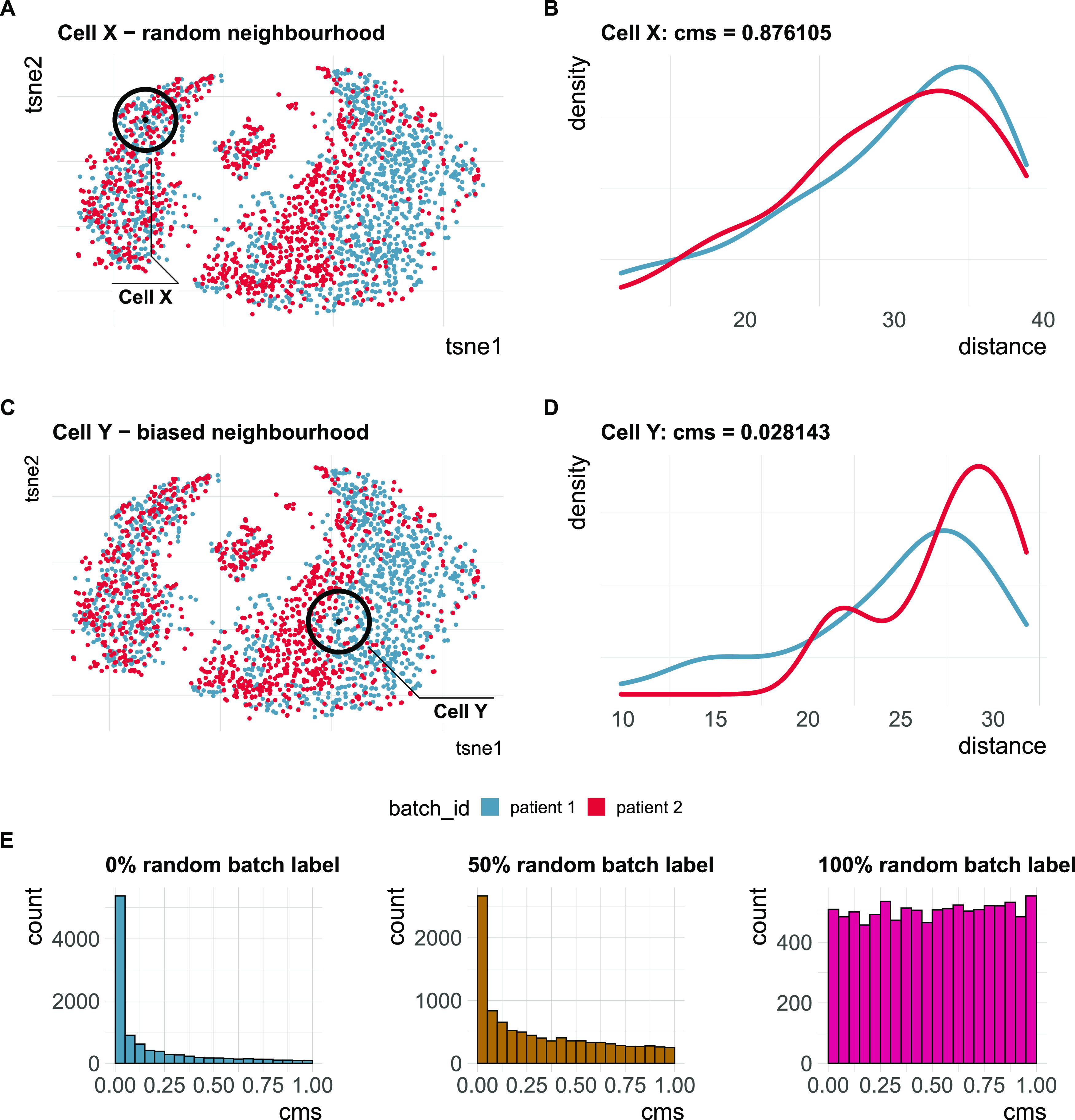
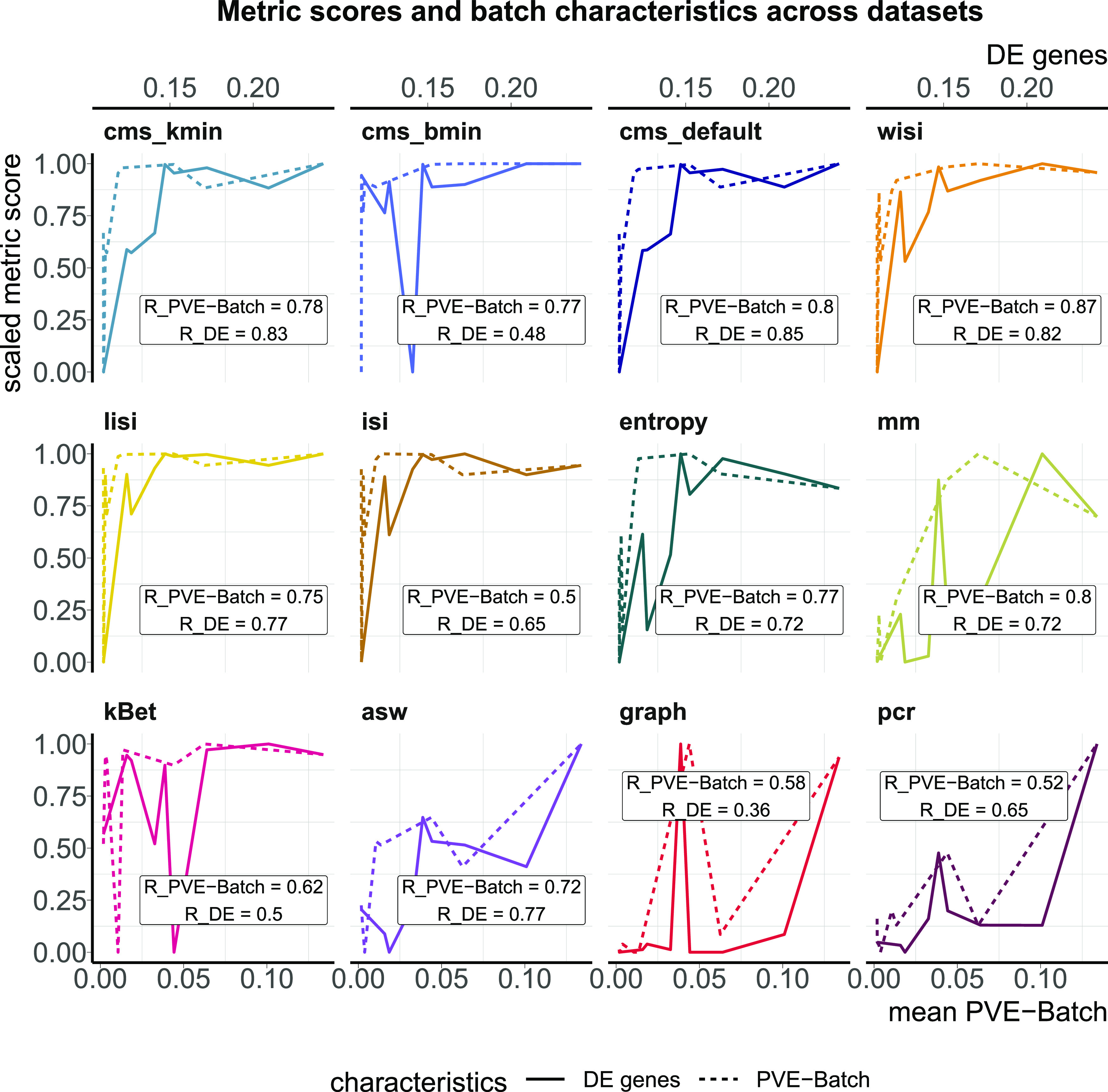
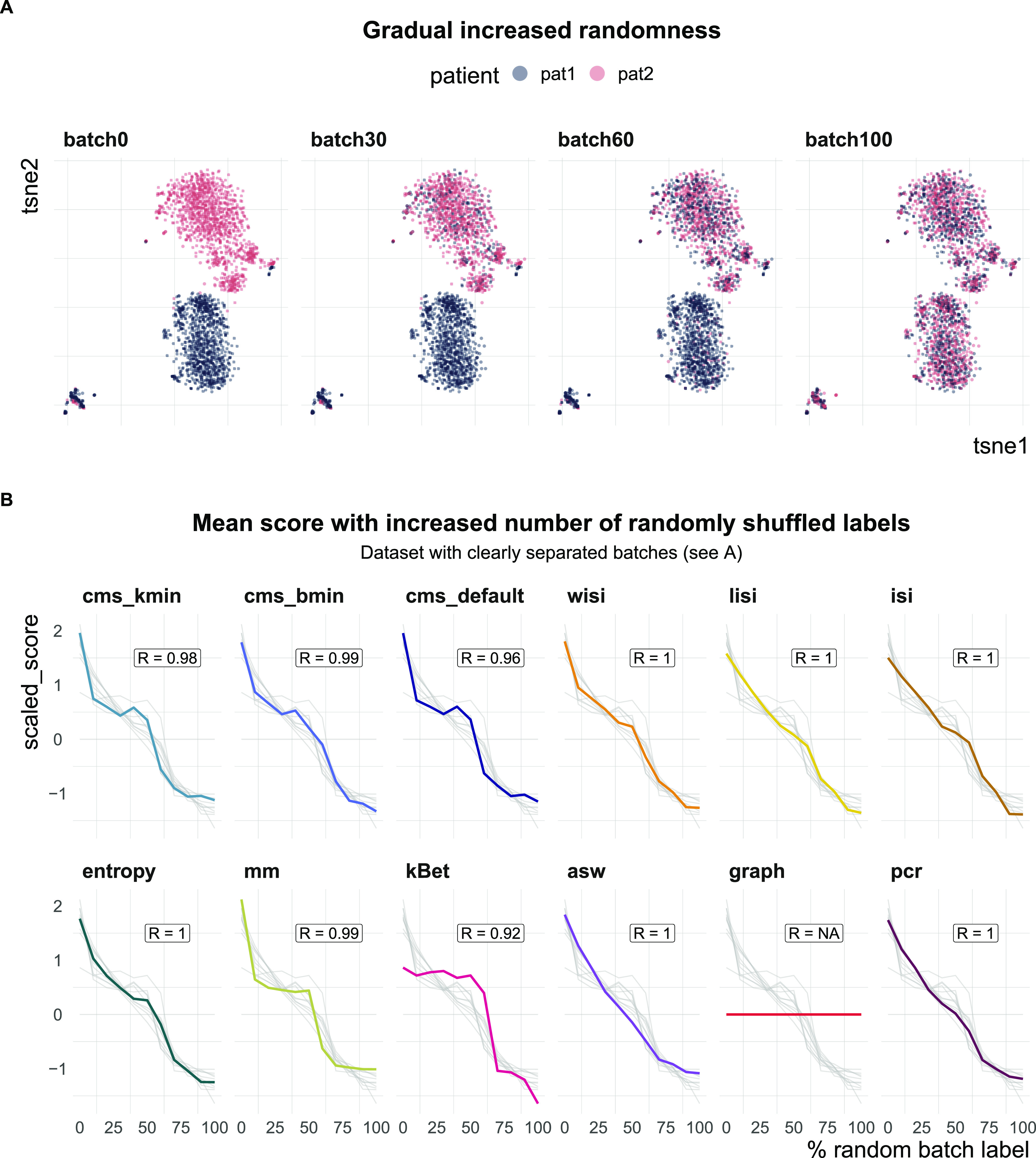
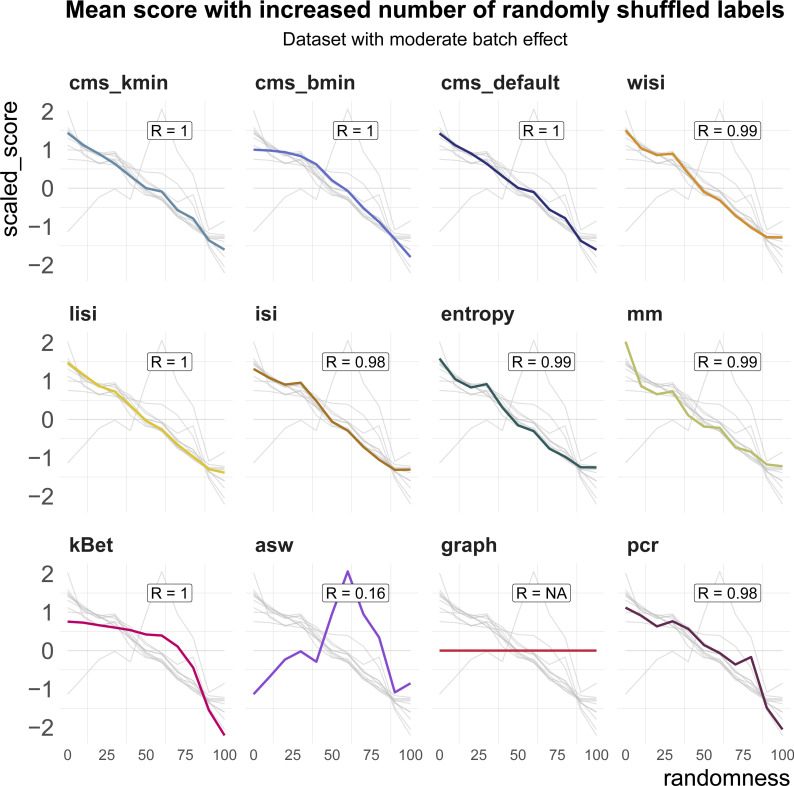
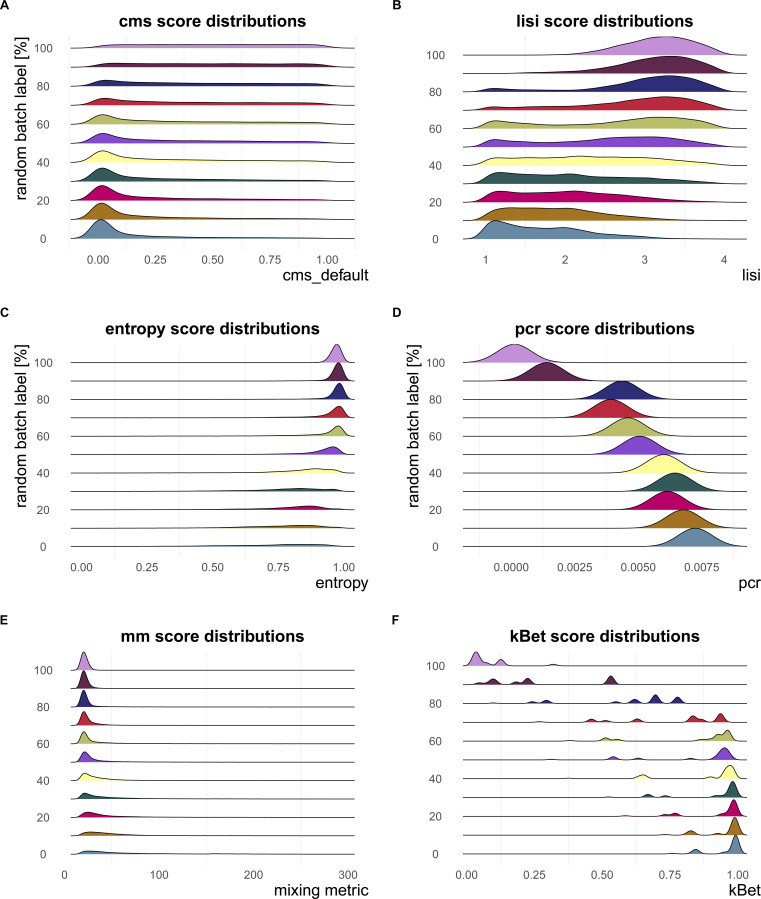
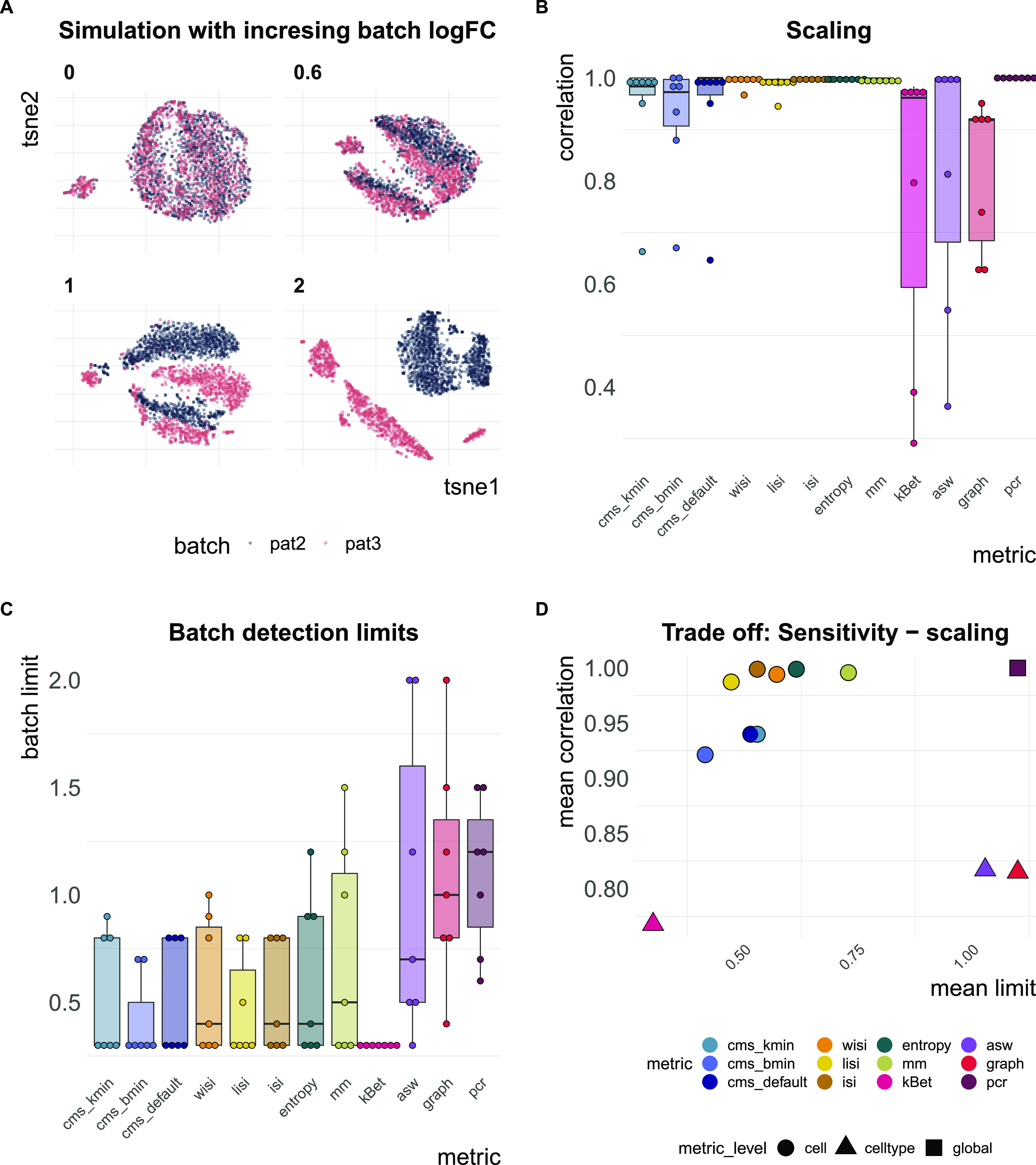
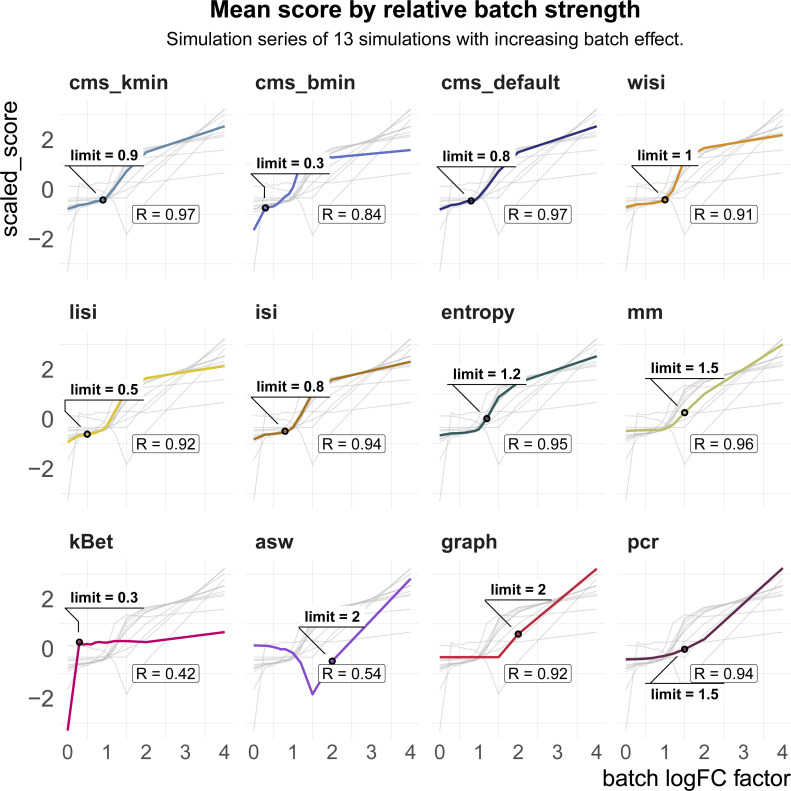
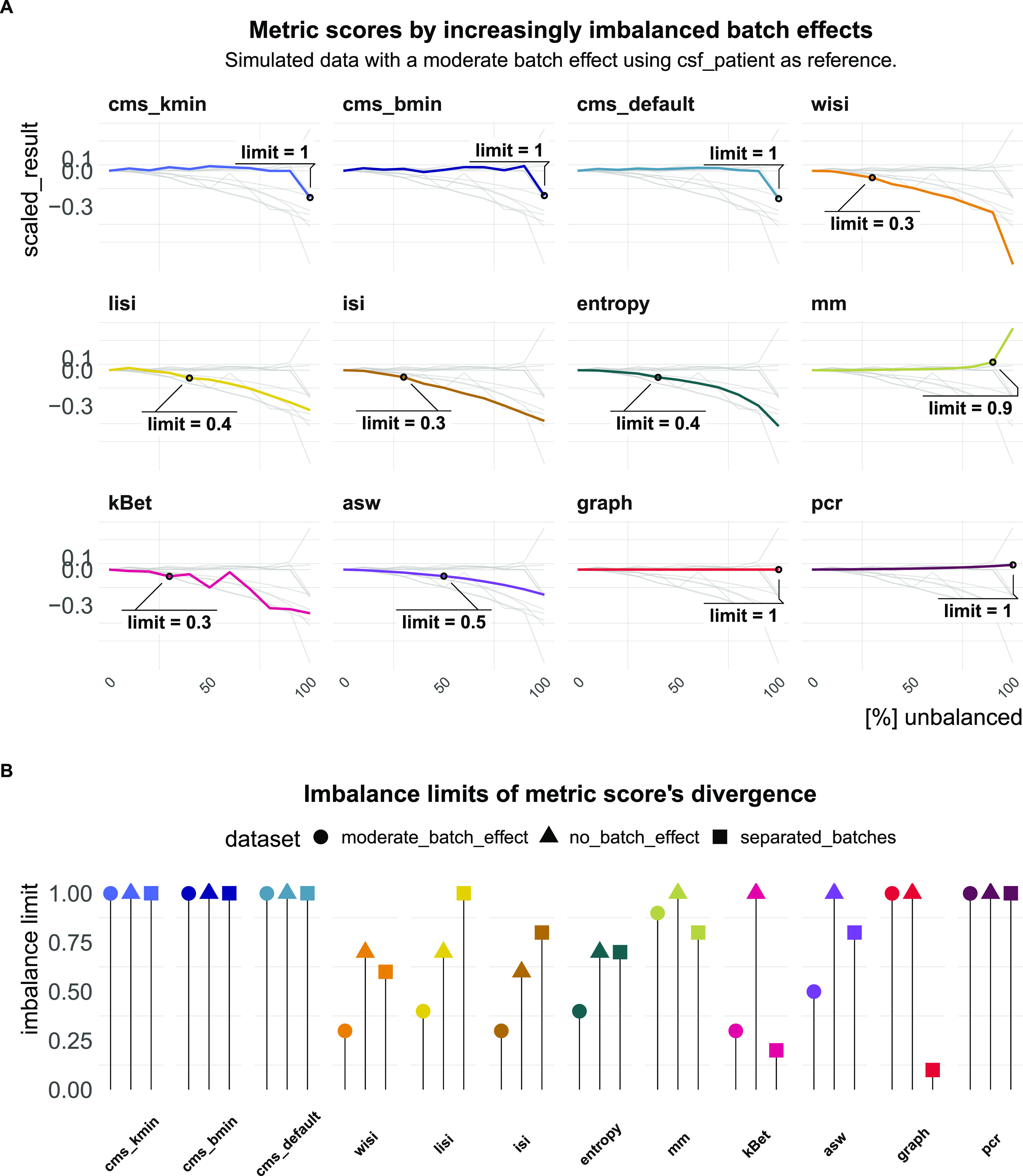
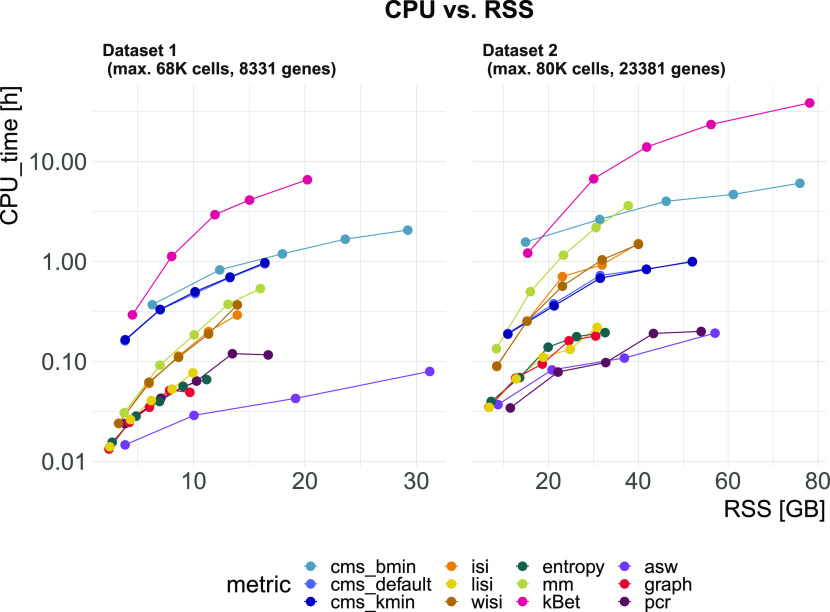
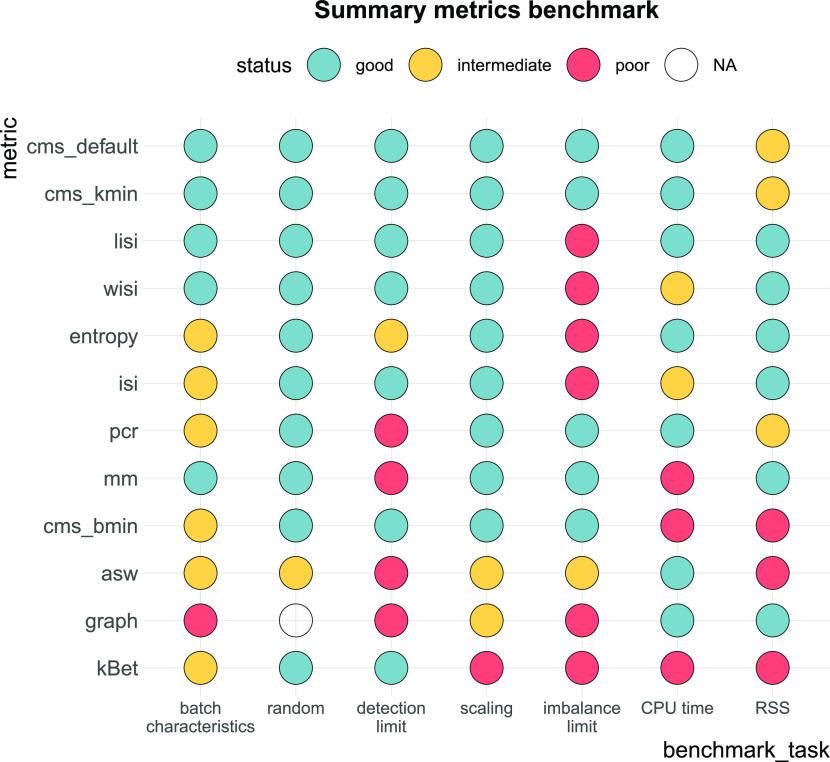
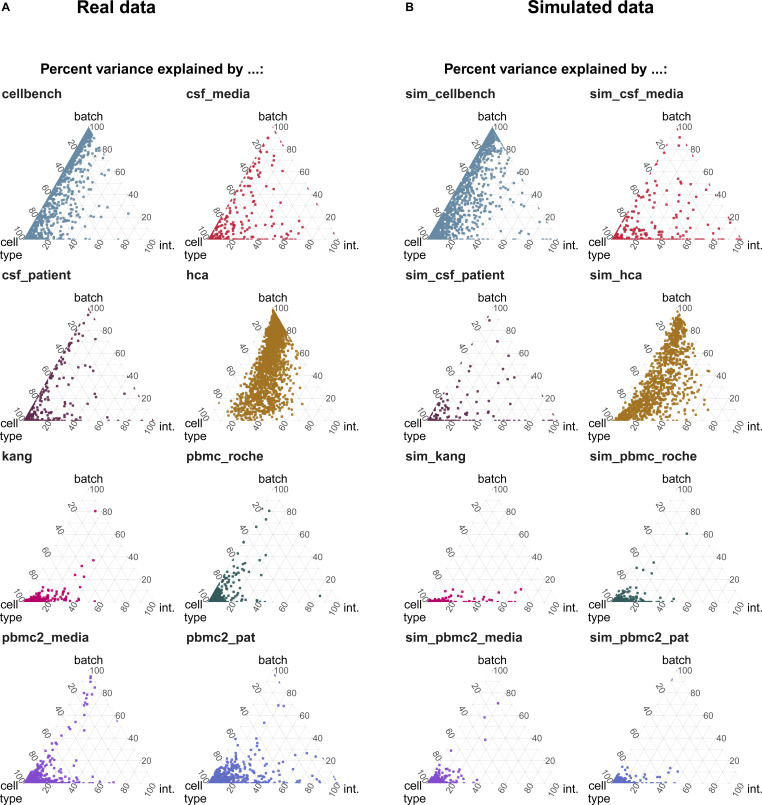
A key challenge in single-cell RNA-sequencing (scRNA-seq) data analysis is batch effects that can obscure the biological signal of interest. Although there are various tools and methods to correct for batch effects, their performance can vary. Therefore, it is important to understand how batch effects manifest to adjust for them. Here, we systematically explore batch effects across various scRNA-seq datasets according to magnitude, cell type specificity, and complexity. We developed a cell-specific mixing score (cms) that quantifies mixing of cells from multiple batches. By considering distance distributions, the score is able to detect local batch bias as well as differentiate between unbalanced batches and systematic differences between cells of the same cell type. We compare metrics in scRNA-seq data using real and synthetic datasets and whereas these metrics target the same question and are used interchangeably, we find differences in scalability, sensitivity, and ability to handle differentially abundant cell types. We find that cell-specific metrics outperform cell type-specific and global metrics and recommend them for both method benchmarks and batch exploration.
单细胞 RNA 测序 (scRNA-seq) 数据分析中的一个关键挑战是批次效应,它可能会掩盖感兴趣的生物学信号。尽管有各种工具和方法可以纠正批次效应,但它们的性能可能会有所不同。因此,了解批次效应的表现方式以进行调整非常重要。在这里,我们根据幅度、细胞类型特异性和复杂性,系统地探索了各种 scRNA-seq 数据集的批次效应。我们开发了一种细胞特异性混合分数 (cms),用于量化来自多个批次的细胞混合情况。通过考虑距离分布,该分数能够检测到局部批次偏差,以及区分不平衡批次和同一细胞类型的细胞之间的系统差异。我们使用真实和合成数据集比较了 scRNA-seq 数据中的指标,尽管这些指标针对的是同一个问题且可以互换使用,但我们发现它们在可扩展性、灵敏度和处理差异丰度细胞类型的能力方面存在差异。我们发现细胞特异性指标优于细胞类型特异性和全局指标,并建议将其用于方法基准测试和批次探索。