Department of Molecular Life Sciences, University of Zurich, Zurich, Switzerland.
SIB Swiss Institute of Bioinformatics, Zurich, Switzerland.
Nat Commun. 2020 Nov 30;11(1):6077. doi: 10.1038/s41467-020-19894-4.
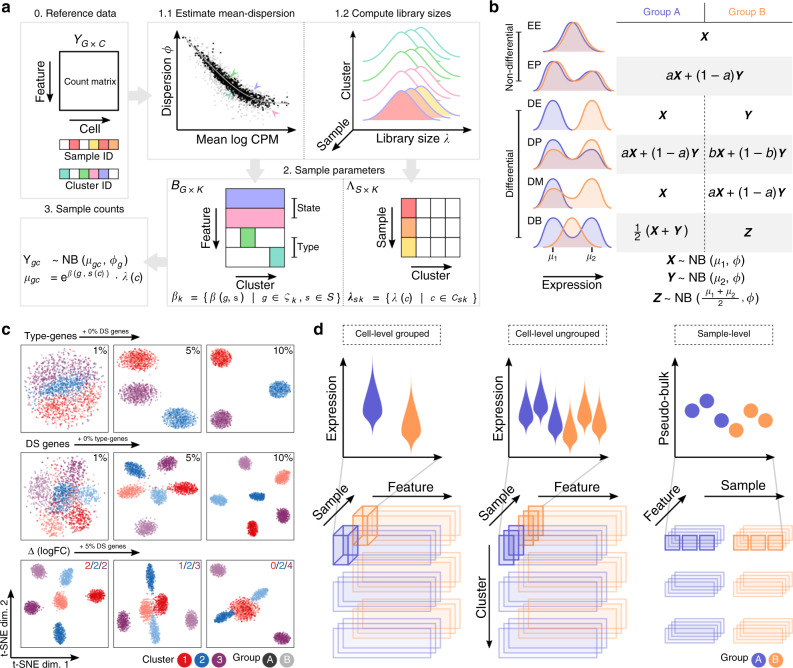
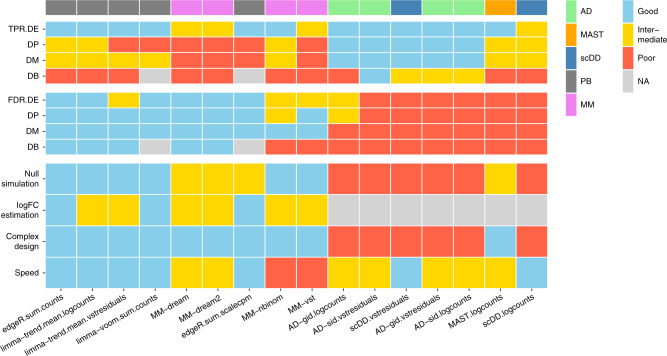
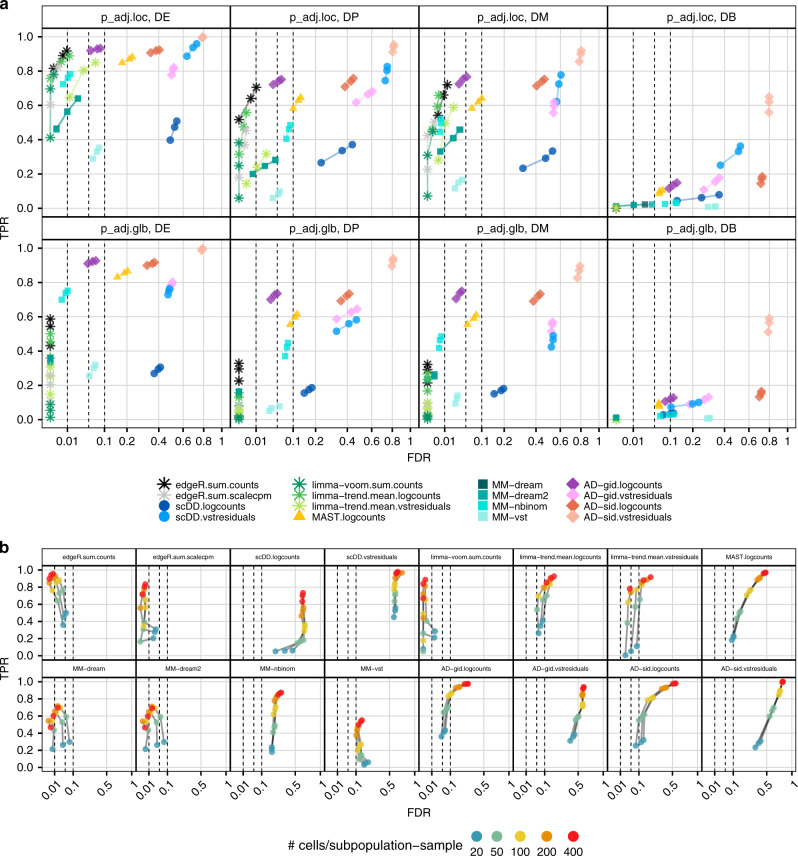
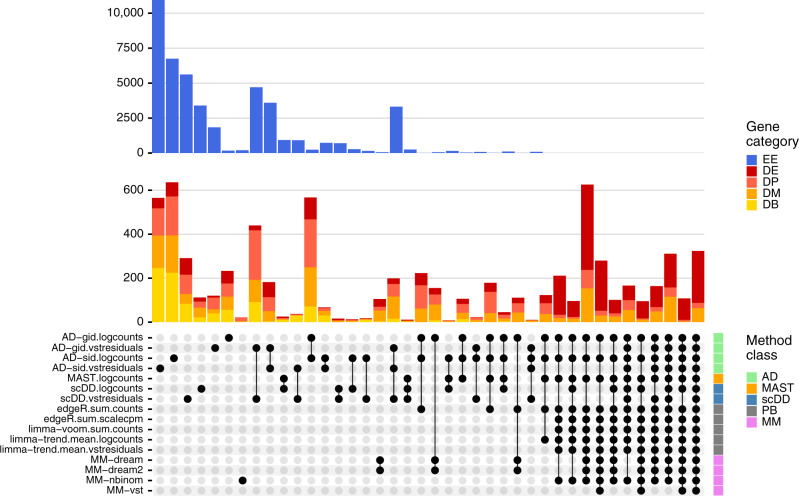
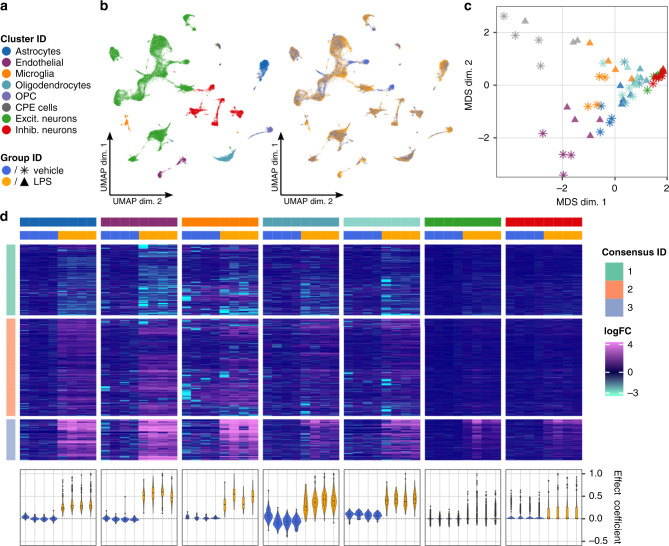
Single-cell RNA sequencing (scRNA-seq) has become an empowering technology to profile the transcriptomes of individual cells on a large scale. Early analyses of differential expression have aimed at identifying differences between subpopulations to identify subpopulation markers. More generally, such methods compare expression levels across sets of cells, thus leading to cross-condition analyses. Given the emergence of replicated multi-condition scRNA-seq datasets, an area of increasing focus is making sample-level inferences, termed here as differential state analysis; however, it is not clear which statistical framework best handles this situation. Here, we surveyed methods to perform cross-condition differential state analyses, including cell-level mixed models and methods based on aggregated pseudobulk data. To evaluate method performance, we developed a flexible simulation that mimics multi-sample scRNA-seq data. We analyzed scRNA-seq data from mouse cortex cells to uncover subpopulation-specific responses to lipopolysaccharide treatment, and provide robust tools for multi-condition analysis within the muscat R package.
单细胞 RNA 测序 (scRNA-seq) 已成为一种强大的技术,可大规模描绘单个细胞的转录组。早期的差异表达分析旨在识别亚群之间的差异,以鉴定亚群标记。更一般地说,这种方法比较了细胞组之间的表达水平,从而导致了跨条件分析。鉴于重复的多条件 scRNA-seq 数据集的出现,一个越来越受到关注的领域是进行样本水平的推断,这里称为差异状态分析;然而,目前尚不清楚哪种统计框架最适合处理这种情况。在这里,我们调查了执行跨条件差异状态分析的方法,包括基于细胞水平的混合模型和基于聚合伪总体数据的方法。为了评估方法的性能,我们开发了一种灵活的模拟,模拟多样本 scRNA-seq 数据。我们分析了来自小鼠大脑皮层细胞的 scRNA-seq 数据,以揭示脂多糖处理对特定亚群的反应,并在 muscat R 包中提供了用于多条件分析的强大工具。