Wang Junling, Liu Zhimei, Xu Manting, Han Xiaodi, Ren Changhong, Yang Xinying, Zhang Chunhua, Fang Fang
Department of Neurology, Beijing Children's Hospital, Capital Medical University, National Center for Children's Health, Beijing, China.
Department of Research, Development of MILS International, Ishikawa, Japan.
Front Pharmacol. 2021 Mar 8;12:605803. doi: 10.3389/fphar.2021.605803. eCollection 2021.
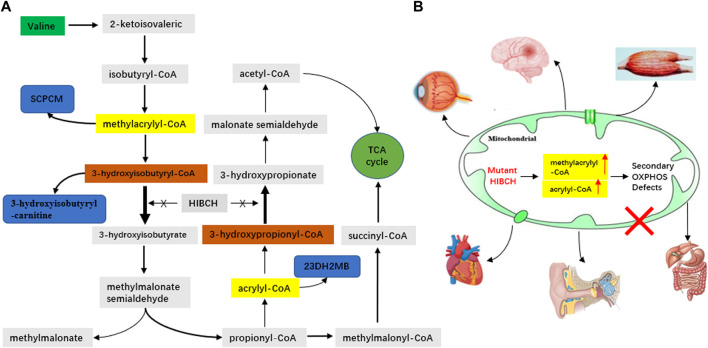
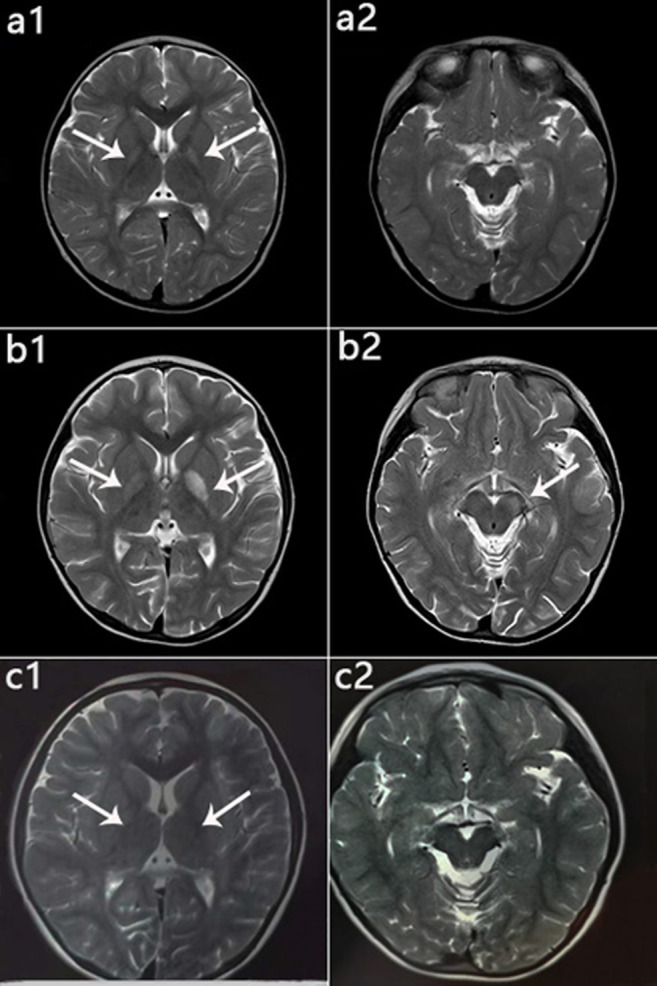
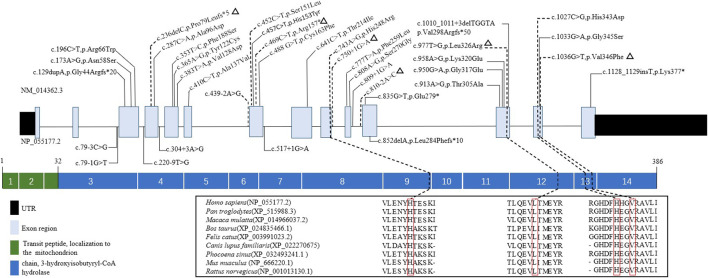
3-Hydroxyisobutyryl-CoA hydrolase (, NM_014362.3) gene mutation can cause HIBCH deficiency, leading to Leigh/Leigh-like disease. To date, few case series have investigated the relationship between metabolites and clinical phenotypes or the effects of treatment, although 34 patients with mutations from 27 families have been reported. The purpose of this study was to analyze the phenotypic spectrum, follow-up results, metabolites, and genotypes of patients with HIBCH deficiency presenting with Leigh/Leigh-like syndrome and explore specific metabolites related to disease diagnosis and prognosis through retrospective and longitudinal studies. Applying next-generation sequencing, we identified eight patients with mutations from our cohort of 181 cases of genetically diagnosed Leigh/Leigh-like syndrome. Six novel mutations were identified: c.977T>G [p.Leu326Arg], c.1036G>T [p.Val346Phe], c.750+1G>A, c.810-2A>C, c.469C>T [p.Arg157*], and c.236delC [p.Pro79Leufs*5]. The Newcastle Pediatric Mitochondrial Disease Scale (NPMDS) was employed to assess disease progression and clinical outcomes. The non-invasive approach of metabolite analysis showed that levels of some were associated with clinical phenotype severity. Five (5/7) patients presented with elevated C4-OH in dried blood spots, and the level was probably correlated with the NPMDS scores during the peak disease phase. 2,3-Dihydroxy-2-methylbutyrate in urine was elevated in six (6/7) patients and elevated S-(2-caboxypropyl)cysteamine in urine was found in three patients (3/3). The median age at initial presentation was 13 months (8-18 months), and the median follow-up was 2.3 years (range 1.3-7.2 years). We summarized and compared with all reported patients with mutations. The most prominent clinical manifestations were developmental regression/delay, hypotonia, encephalopathy, and feeding difficulties. We administered drug and dietary treatment. During follow-up, five patients responded positively to treatment with a significant decrease in NPMDS scores. Our research is the largest case series of patients with mutations.
3-羟基异丁酰辅酶A水解酶(,NM_014362.3)基因突变可导致3-羟基异丁酰辅酶A水解酶缺乏症,进而引发 Leigh 综合征/Leigh 样疾病。迄今为止,尽管已有报道来自27个家庭的34例携带该基因突变的患者,但很少有病例系列研究代谢物与临床表型之间的关系或治疗效果。本研究的目的是通过回顾性和纵向研究,分析表现为 Leigh 综合征/Leigh 样综合征的3-羟基异丁酰辅酶A水解酶缺乏症患者的表型谱、随访结果、代谢物和基因型,并探索与疾病诊断和预后相关的特定代谢物。应用二代测序技术,我们在181例经基因诊断为 Leigh 综合征/Leigh 样综合征的队列中,鉴定出8例携带该基因突变的患者。鉴定出6个新的该基因突变:c.977T>G [p.Leu326Arg]、c.1036G>T [p.Val346Phe]、c.750+1G>A、c.810-2A>C、c.469C>T [p.Arg157*]和 c.236delC [p.Pro79Leufs*5]。采用纽卡斯尔儿童线粒体疾病量表(NPMDS)评估疾病进展和临床结局。代谢物分析的非侵入性方法显示,某些代谢物水平与临床表型严重程度相关。7例患者中有5例干血斑中的 C4-OH 升高,且该水平可能与疾病高峰期的 NPMDS 评分相关。7例患者中有6例尿中的2,3-二羟基-2-甲基丁酸升高,3例患者(3/3)尿中的 S-(2-羧丙基)半胱胺升高。首次就诊时的中位年龄为13个月(8 - 18个月),中位随访时间为2.3年(范围1.3 - 7.2年)。我们总结并与所有已报道的携带该基因突变的患者进行了比较。最突出的临床表现为发育倒退/延迟、肌张力减退、脑病和喂养困难。我们给予了药物和饮食治疗。随访期间,5例患者对治疗反应良好,NPMDS 评分显著降低。我们的研究是携带该基因突变患者最大的病例系列。