Department of Endocrinology and Metabolism, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China.
Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China.
Mol Genet Genomic Med. 2021 May;9(5):e1668. doi: 10.1002/mgg3.1668. Epub 2021 Mar 25.
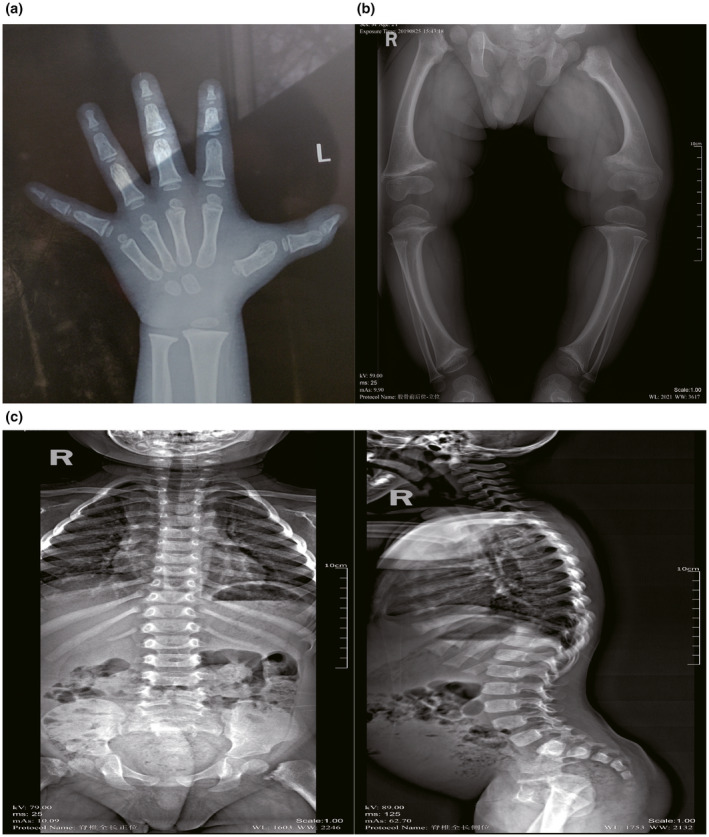
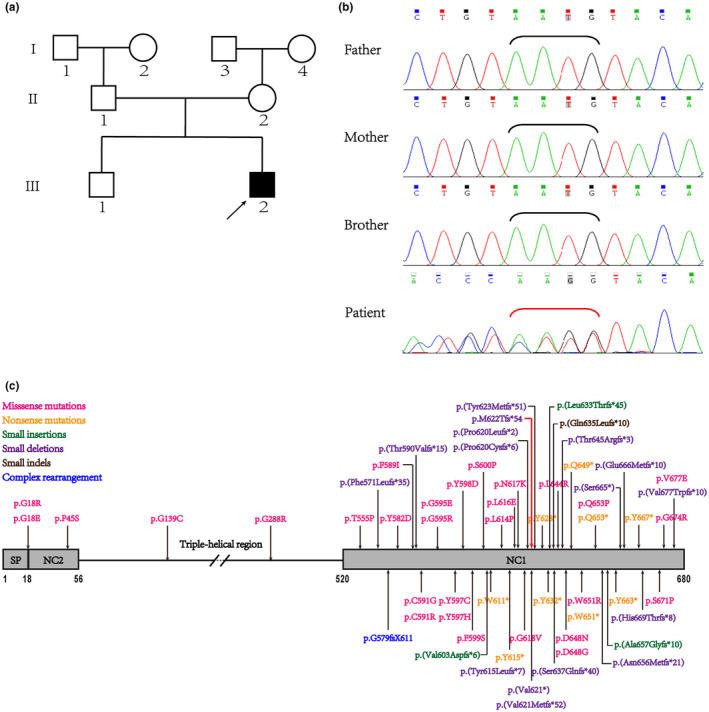
Schmid-type metaphyseal chondrodysplasia (SMCD) is a rare autosomal dominant skeletal dysplasia caused by heterozygous mutations in COL10A1, the gene which encodes collagen type X alpha 1 chain. However, its genotype-phenotype relationship has not been fully determined. Subjects and Methods The proband is a 2-year-old boy, born of non-consanguineous Chinese parents. We conducted a systematic analysis of the clinical and radiological characteristics and a follow-up study of the proband. Whole-exome sequencing was applied for the genetic analysis, together with bioinformatic analysis of predicted consequences of the identified variant. A homotrimer model was built to visualize the affected region and predict possible outcomes of this variant. Furthermore, a literature review and genotype-phenotype analysis were performed by online searching all cases with SMCD.
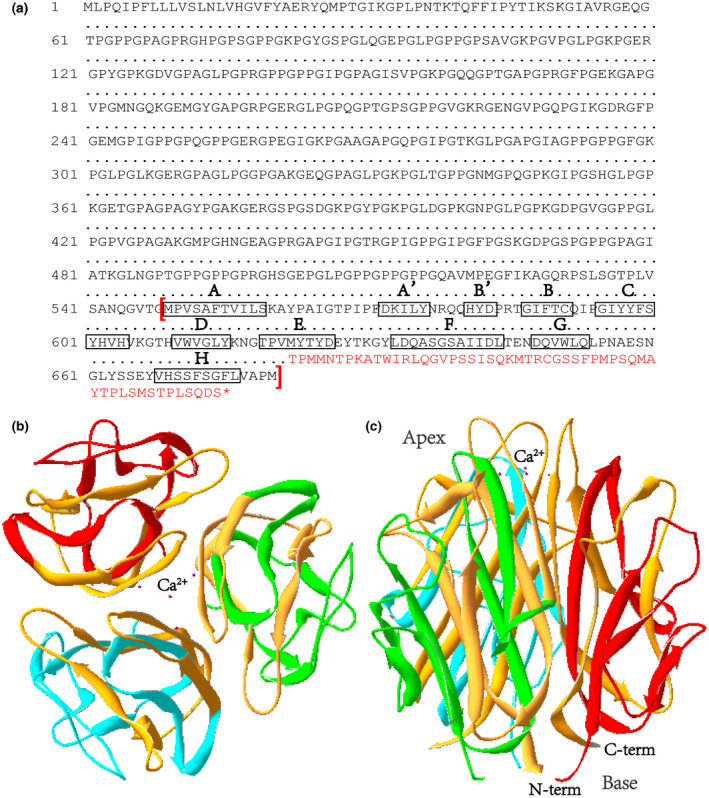
A novel heterozygous variant (NM_000493.4: c.1863_1866delAATG, NP_000484.2: p.(Met622 Thrfs*54)) was identified in COL10A1 gene in the affected child. And it was predicted to be pathogenic by in silico analysis. Protein modeling revealed that the variant was located in the NC1 domain, which was predicted to produce truncated collagen and impair the trimerization of collagen type X alpha 1 chain and combination with molecules in the matrix. Moreover, genotype-phenotype correlation analysis demonstrated that patients with truncating variants or variants in NC1 domain often presented earlier onset and severer symptoms compared with those with non-truncating or variants in non-NC1 domains.
The NC1 domain of COL10A1 was proved to be the hotspot region underlying SMCD, patients with variants in NC1 domain were more likely to present severer manifestations at an earlier age.
Schmid 型干骺端软骨发育不良(SMCD)是一种罕见的常染色体显性遗传性骨骼发育不良,由 COL10A1 基因杂合突变引起,该基因编码胶原 X 型α 1 链。然而,其基因型-表型关系尚未完全确定。
先证者为 2 岁男孩,父母非近亲结婚。我们对其进行了临床和放射学特征的系统分析,并对先证者进行了随访研究。应用全外显子组测序进行遗传分析,并对鉴定出的变异进行生物信息学分析。构建同源三聚体模型,以可视化受影响区域并预测该变异的可能结果。此外,通过在线搜索所有 SMCD 病例进行文献复习和基因型-表型分析。
在受影响的孩子中发现 COL10A1 基因中的一个新的杂合变异(NM_000493.4:c.1863_1866delAATG,NP_000484.2:p.(Met622Thrfs*54))。通过计算机分析预测该变异具有致病性。蛋白质建模表明,该变异位于 NC1 结构域,预计会产生截短的胶原蛋白,从而破坏胶原蛋白 X 型α 1 链的三聚体形成以及与基质中分子的结合。此外,基因型-表型相关性分析表明,与非截短或非 NC1 结构域变异的患者相比,截断变异或 NC1 结构域变异的患者发病更早,症状更严重。
COL10A1 的 NC1 结构域被证实是 SMCD 的热点区域,NC1 结构域变异的患者更有可能在更早的年龄出现更严重的表现。