Kim Heon Seok, Hwang Gue-Ho, Lee Hyomin K, Bae Taegeun, Park Seong-Ho, Kim Yong Jun, Lee Sun, Park Jae-Hoon, Bae Sangsu, Hur Junho K
Division of Oncology, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA.
Department of Chemistry, Hanyang University, Seoul 04763, South Korea.
Mol Ther Methods Clin Dev. 2021 Jan 11;20:792-800. doi: 10.1016/j.omtm.2020.10.012. eCollection 2021 Mar 12.
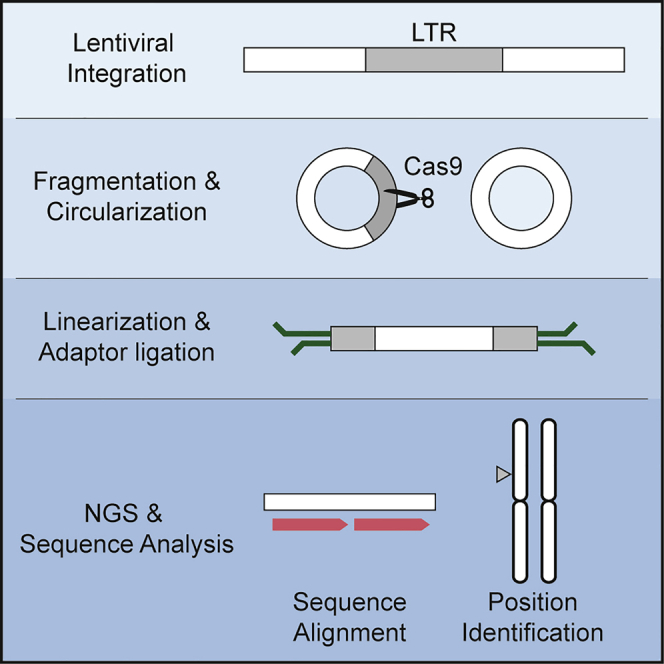
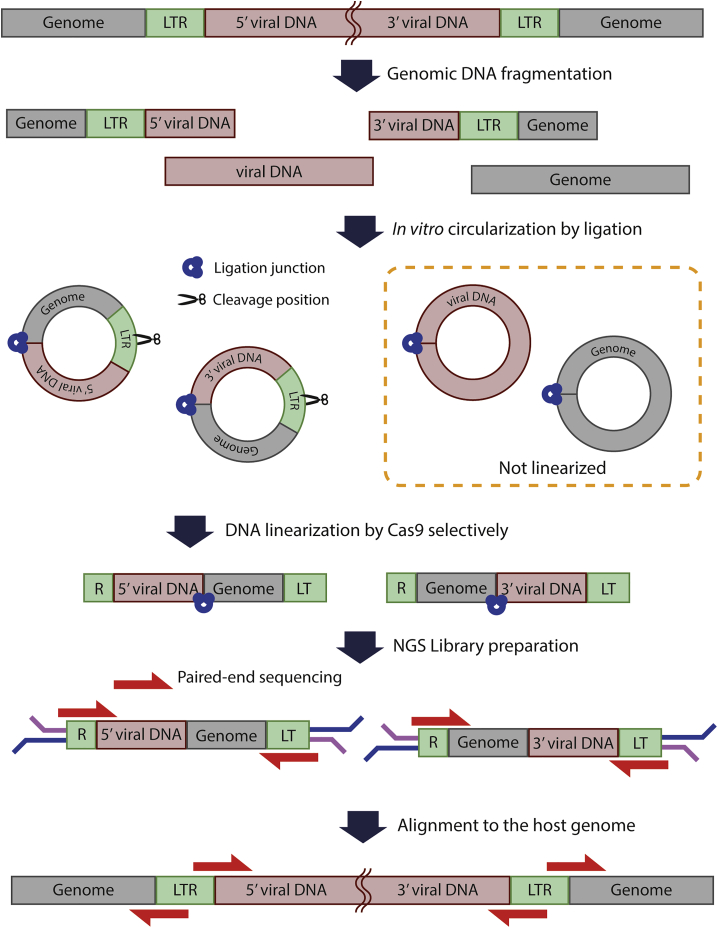
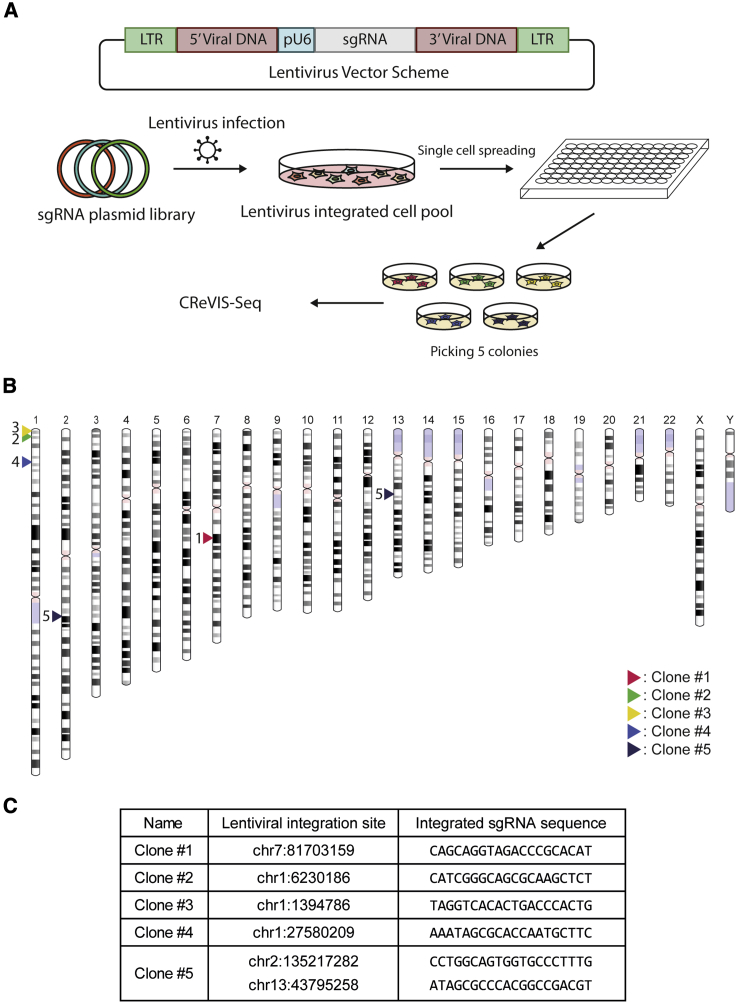
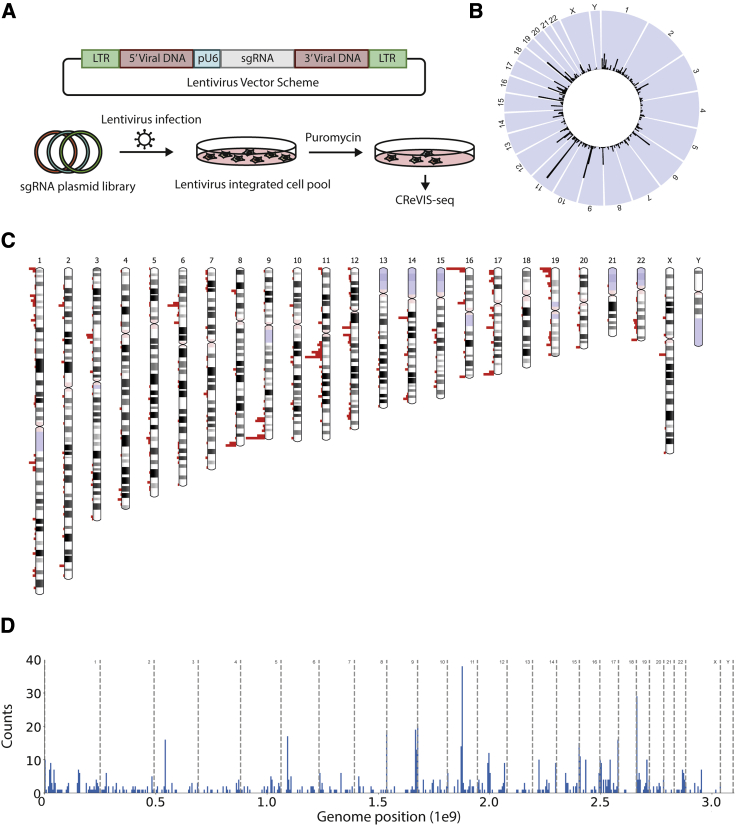
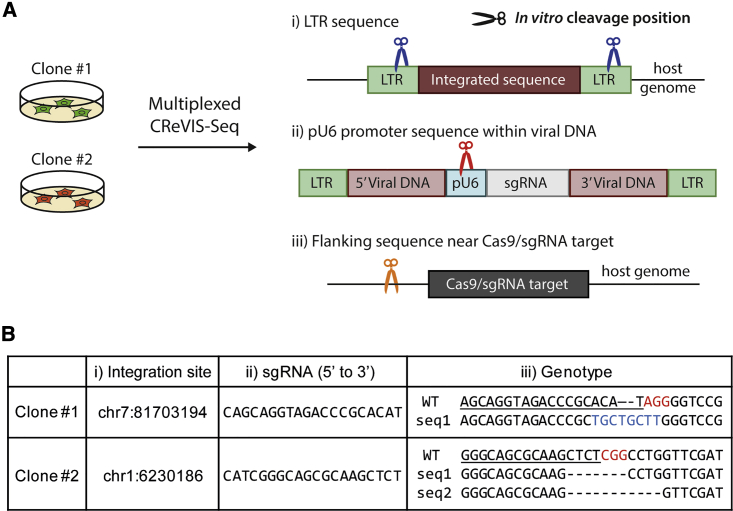
Lentiviruses have been widely used as a means of transferring exogenous DNAs into human cells to treat various genetic diseases. Lentiviral vectors are fundamentally integrated into the host genome, but their integration sites are generally unpredictable, which may increase the uncertainty for their use in therapeutics. To determine the viral integration sites in the host genome, several PCR-based methods have been developed. However, the sensitivities of the PCR-based methods are highly dependent on the primer sequences, and optimized primer design is required for individual target sites. In order to address this issue, we developed an alternative method for genome-wide mapping of viral insertion sites, named CReVIS-seq (CRISPR-enhanced Viral Integration Site Sequencing). The method is based on the sequential steps: fragmentation of genomic DNAs, circularization, cleavage of target sequence in a CRISPR guide RNA-specific manner, high-throughput sequencing of the linearized DNA fragments in an unbiased manner, and identification of viral insertion sites via sequence analysis. By design, CReVIS-seq is not affected by biases that could be introduced during the target enrichment step via PCR amplification using site specific primers. Furthermore, we found that multiplexed CReVIS-seq, using collections of different single-guide RNAs (sgRNAs), enables simultaneous identification of multiple target sites and structural variations (i.e., circularized viral genome), in both single cell clones and heterogeneous cell populations.
慢病毒已被广泛用作将外源DNA导入人类细胞以治疗各种遗传疾病的手段。慢病毒载体基本上会整合到宿主基因组中,但其整合位点通常不可预测,这可能会增加其在治疗应用中的不确定性。为了确定宿主基因组中的病毒整合位点,已经开发了几种基于PCR的方法。然而,基于PCR的方法的灵敏度高度依赖于引物序列,并且需要针对各个靶位点进行优化的引物设计。为了解决这个问题,我们开发了一种用于全基因组病毒插入位点定位的替代方法,称为CReVIS-seq(CRISPR增强的病毒整合位点测序)。该方法基于以下连续步骤:基因组DNA片段化、环化、以CRISPR引导RNA特异性方式切割靶序列、以无偏方式对线性化DNA片段进行高通量测序,以及通过序列分析鉴定病毒插入位点。通过设计,CReVIS-seq不受在靶富集步骤中使用位点特异性引物进行PCR扩增时可能引入的偏差的影响。此外,我们发现使用不同单引导RNA(sgRNA)集合的多重CReVIS-seq能够在单细胞克隆和异质细胞群体中同时鉴定多个靶位点和结构变异(即环化病毒基因组)。