Department of Pathology and Biomedical Science, University of Otago, Christchurch, New Zealand.
Department of Biochemistry, University of Otago, Dunedin, New Zealand.
Sci Rep. 2021 Mar 30;11(1):7192. doi: 10.1038/s41598-021-86690-5.
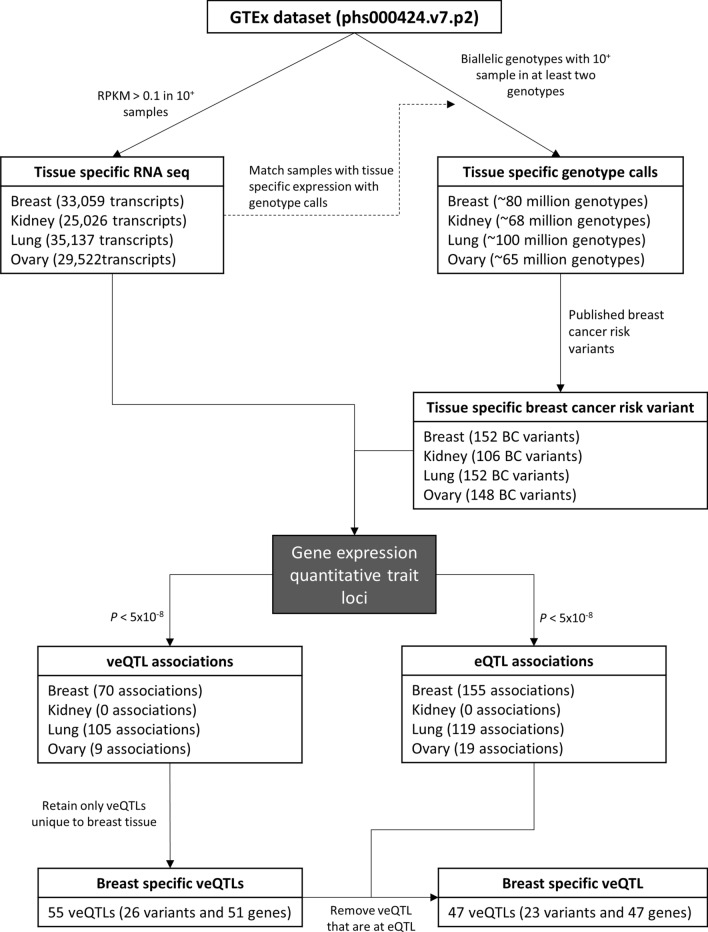
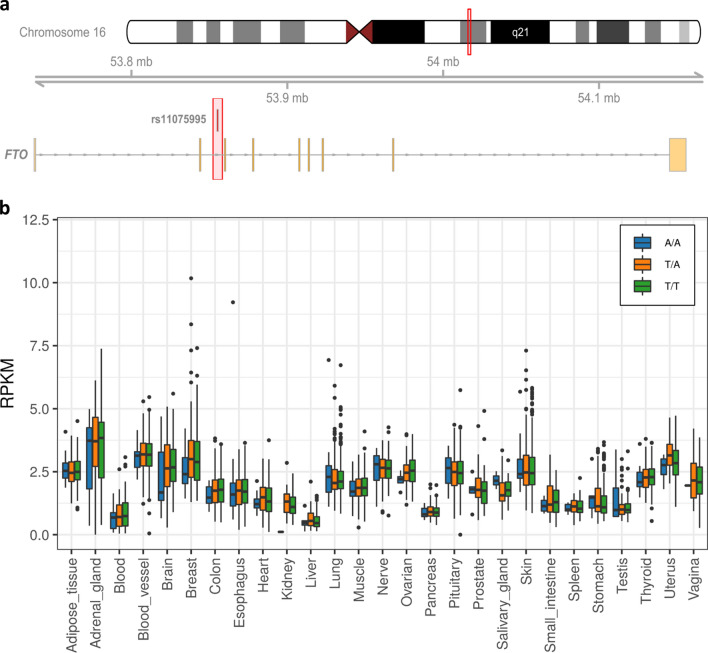
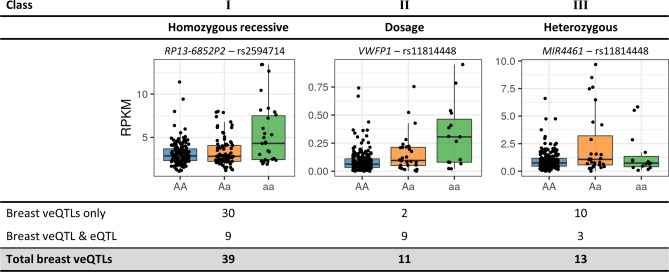
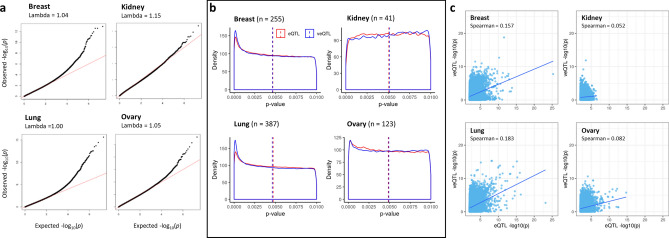
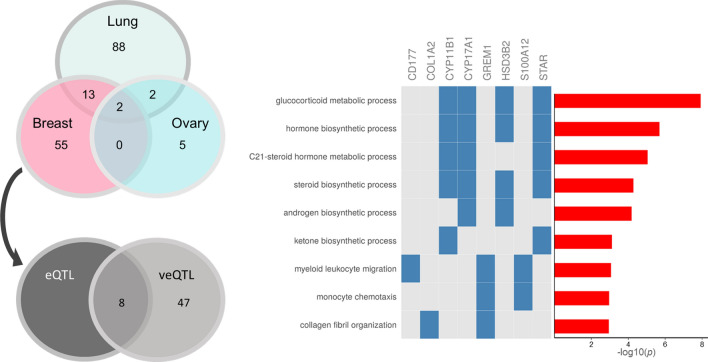
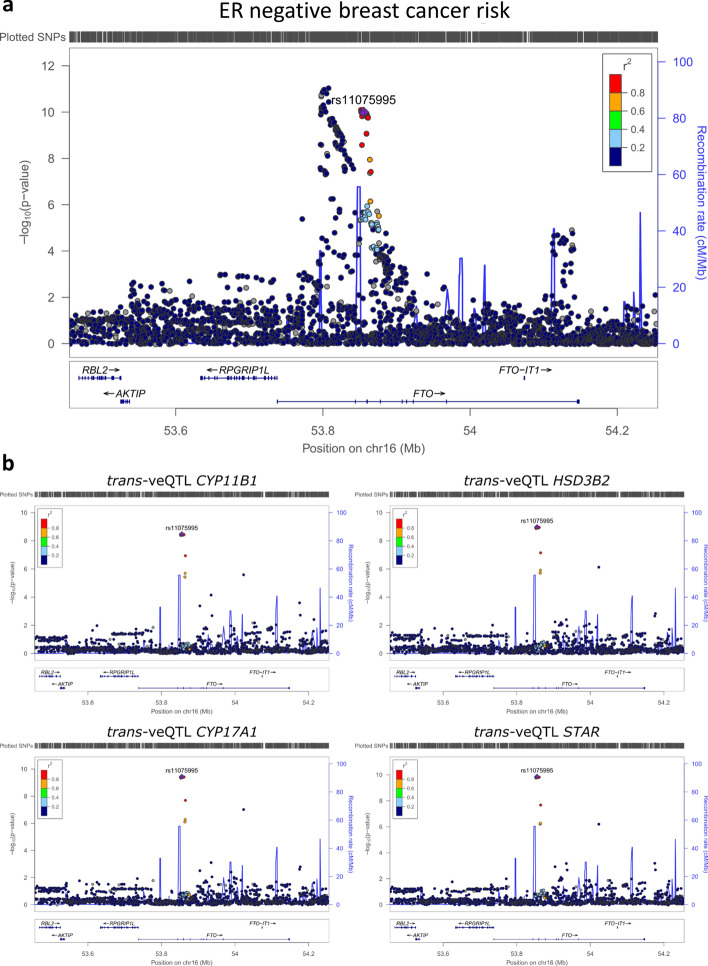
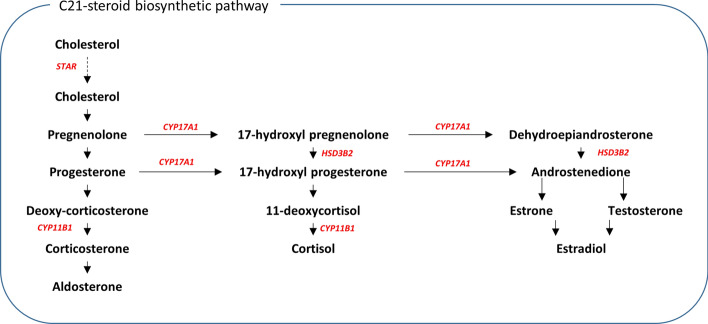
Genome wide association studies (GWAS) have identified more than 180 variants associated with breast cancer risk, however the underlying functional mechanisms and biological pathways which confer disease susceptibility remain largely unknown. As gene expression traits are under genetic regulation we hypothesise that differences in gene expression variability may identify causal breast cancer susceptibility genes. We performed variable expression quantitative trait loci (veQTL) analysis using tissue-specific expression data from the Genotype-Tissue Expression (GTEx) Common Fund Project. veQTL analysis identified 70 associations (p < 5 × 10) consisting of 60 genes and 27 breast cancer risk variants, including 55 veQTL that were observed in breast tissue only. Pathway analysis of genes associated with breast-specific veQTL revealed an enrichment of four genes (CYP11B1, CYP17A1 HSD3B2 and STAR) involved in the C21-steroidal biosynthesis pathway that converts cholesterol to breast-related hormones (e.g. oestrogen). Each of these four genes were significantly more variable in individuals homozygous for rs11075995 (A/A) breast cancer risk allele located in the FTO gene, which encodes an RNA demethylase. The A/A allele was also found associated with reduced expression of FTO, suggesting an epi-transcriptomic mechanism may underlie the dysregulation of genes involved in hormonal biosynthesis leading to an increased risk of breast cancer. These findings provide evidence that genetic variants govern high levels of expression variance in breast tissue, thus building a more comprehensive insight into the underlying biology of breast cancer risk loci.
全基因组关联研究(GWAS)已经确定了 180 多个与乳腺癌风险相关的变异,然而,赋予疾病易感性的潜在功能机制和生物学途径在很大程度上仍然未知。由于基因表达特征受遗传调控,我们假设基因表达变异性的差异可能确定乳腺癌易感基因的因果关系。我们使用来自基因型-组织表达(GTEx)共同基金项目的组织特异性表达数据进行了可变表达数量性状基因座(veQTL)分析。veQTL 分析确定了 70 个关联(p < 5×10),由 60 个基因和 27 个乳腺癌风险变异组成,其中 55 个仅在乳腺组织中观察到 veQTL。与乳腺特异性 veQTL 相关的基因的通路分析显示,四个基因(CYP11B1、CYP17A1 HSD3B2 和 STAR)参与 C21-甾体生物合成途径的富集,该途径将胆固醇转化为乳腺相关激素(如雌激素)。位于 FTO 基因中的 rs11075995(A/A)乳腺癌风险等位基因纯合子个体中,这四个基因的每个基因都显著更具变异性,该基因编码一种 RNA 去甲基酶。还发现 A/A 等位基因与 FTO 表达降低相关,这表明 epi 转录组学机制可能是激素生物合成中涉及的基因失调导致乳腺癌风险增加的基础。这些发现为遗传变异控制乳腺组织中高水平表达变异提供了证据,从而更全面地了解乳腺癌风险基因座的潜在生物学。