Cintesis@Rise, Universidade do Algarve, Faro, Portugal.
Centro de Ciências do Mar (CCMAR), Universidade do Algarve, Faro, Portugal.
Sci Rep. 2024 Sep 28;14(1):22526. doi: 10.1038/s41598-024-72163-y.
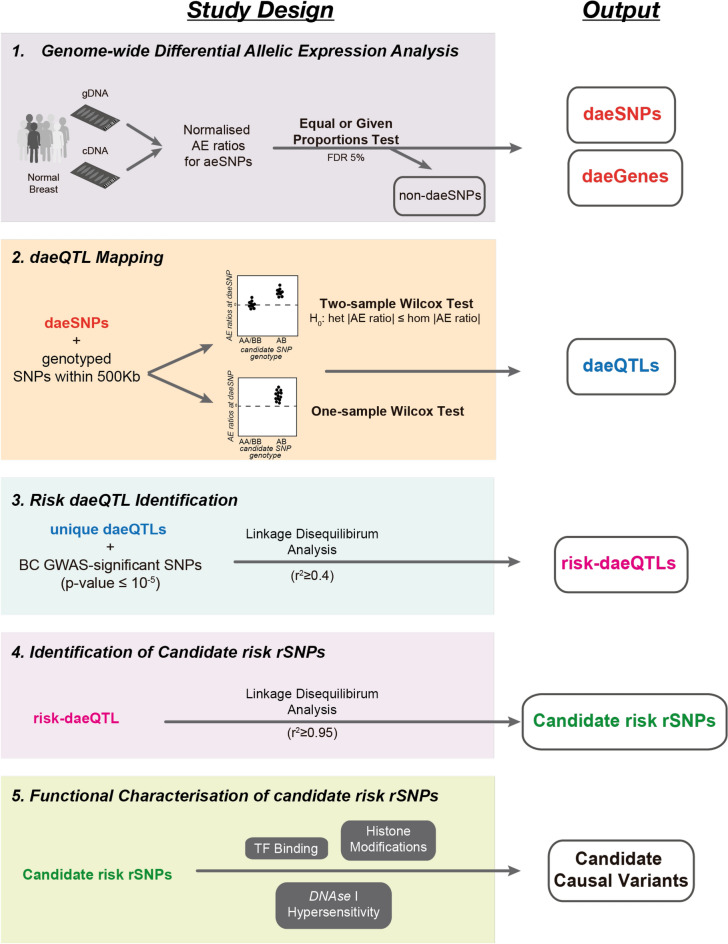
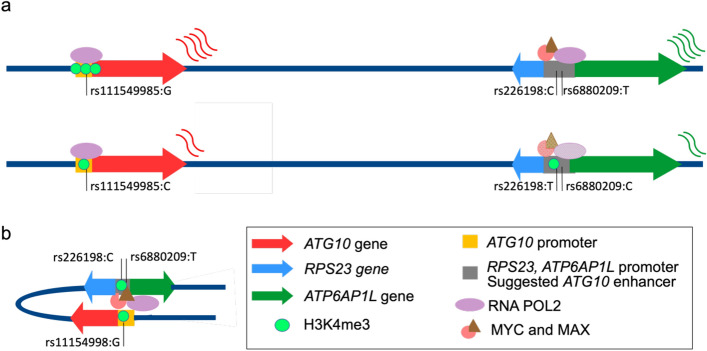
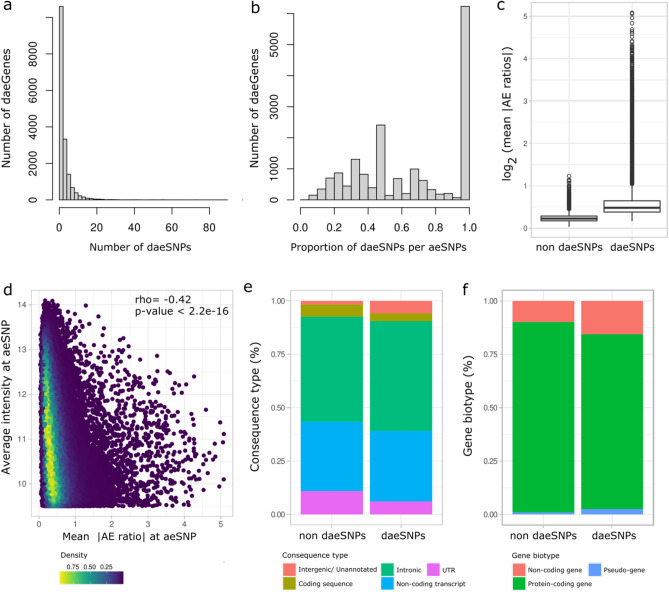
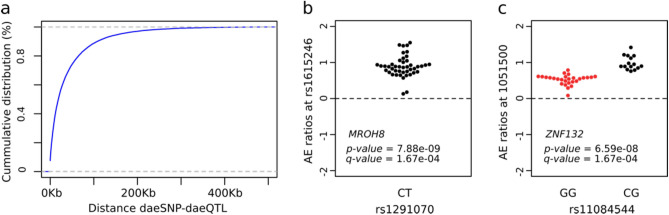
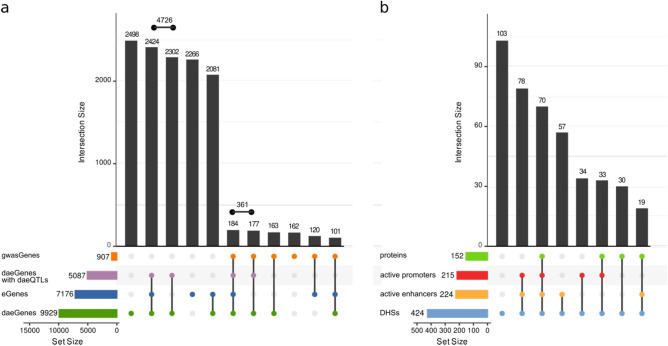
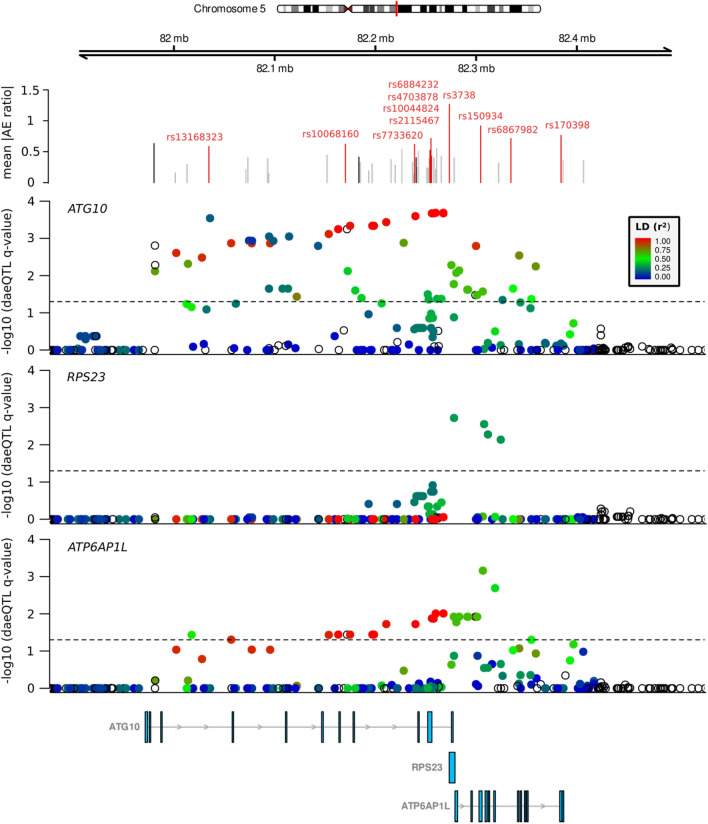
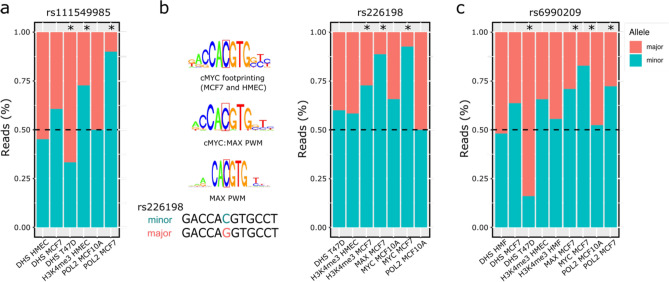
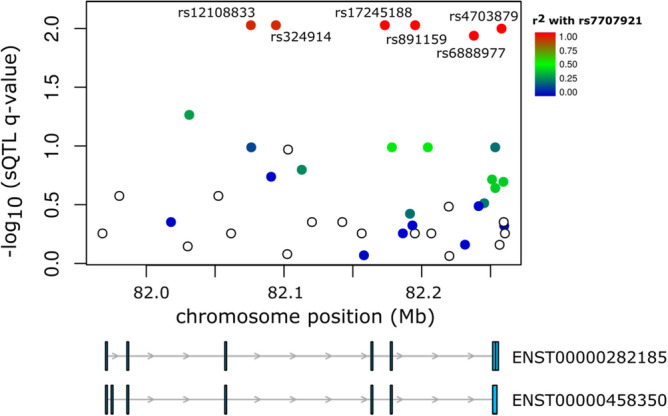
Understanding breast cancer genetic risk relies on identifying causal variants and candidate target genes in risk loci identified by genome-wide association studies (GWAS), which remains challenging. Since most loci fall in active gene regulatory regions, we developed a novel approach facilitated by pinpointing the variants with greater regulatory potential in the disease's tissue of origin. Through genome-wide differential allelic expression (DAE) analysis, using microarray data from 64 normal breast tissue samples, we mapped the variants associated with DAE (daeQTLs). Then, we intersected these with GWAS data to reveal candidate risk regulatory variants and analysed their cis-acting regulatory potential. Finally, we validated our approach by extensive functional analysis of the 5q14.1 breast cancer risk locus. We observed widespread gene expression regulation by cis-acting variants in breast tissue, with 65% of coding and noncoding expressed genes displaying DAE (daeGenes). We identified over 54 K daeQTLs for 6761 (26%) daeGenes, including 385 daeGenes harbouring variants previously associated with BC risk. We found 1431 daeQTLs mapped to 93 different loci in strong linkage disequilibrium with risk-associated variants (risk-daeQTLs), suggesting a link between risk-causing variants and cis-regulation. There were 122 risk-daeQTL with stronger cis-acting potential in active regulatory regions with protein binding evidence. These variants mapped to 41 risk loci, of which 29 had no previous report of target genes and were candidates for regulating the expression levels of 65 genes. As validation, we identified and functionally characterised five candidate causal variants at the 5q14.1 risk locus targeting the ATG10 and ATP6AP1L genes, likely acting via modulation of alternative transcription and transcription factor binding. Our study demonstrates the power of DAE analysis and daeQTL mapping to identify causal regulatory variants and target genes at breast cancer risk loci, including those with complex regulatory landscapes. It additionally provides a genome-wide resource of variants associated with DAE for future functional studies.
理解乳腺癌遗传风险依赖于识别全基因组关联研究(GWAS)确定的风险基因座中的因果变异和候选靶基因,这仍然具有挑战性。由于大多数基因座位于活跃的基因调控区域,我们开发了一种新方法,通过精确定位疾病起源组织中具有更大调控潜力的变异来实现。通过使用来自 64 个正常乳腺组织样本的微阵列数据进行全基因组差异等位基因表达(DAE)分析,我们绘制了与 DAE 相关的变异(daeQTLs)。然后,我们将这些与 GWAS 数据交叉,以揭示候选风险调节变异,并分析它们的顺式作用调节潜力。最后,我们通过对 5q14.1 乳腺癌风险基因座的广泛功能分析验证了我们的方法。我们观察到在乳腺组织中,顺式作用变异广泛调节基因表达,65%的编码和非编码表达基因显示 DAE(daeGenes)。我们鉴定了 54,000 多个 daeQTL 用于 6761 个(26%)daeGenes,包括 385 个携带先前与 BC 风险相关的变异的 daeGenes。我们发现 1431 个 daeQTL 映射到与风险相关变异(风险-daeQTLs)强连锁不平衡的 93 个不同基因座,表明风险引起的变异与顺式调节之间存在联系。有 122 个风险-daeQTL 具有更强的顺式作用潜力,位于具有蛋白质结合证据的活性调控区域。这些变异映射到 41 个风险基因座,其中 29 个以前没有报道过靶基因,是调节 65 个基因表达水平的候选基因。作为验证,我们在 5q14.1 风险基因座鉴定并功能表征了五个候选因果变异,这些变异靶向 ATG10 和 ATP6AP1L 基因,可能通过调节选择性转录和转录因子结合来发挥作用。我们的研究表明,DAE 分析和 daeQTL 映射在识别乳腺癌风险基因座中的因果调节变异和靶基因方面具有强大的功能,包括那些具有复杂调控景观的基因座。此外,它还为未来的功能研究提供了与 DAE 相关的全基因组变异资源。