Tsuru Shota, Vidal Marta L, Pápai Mátyás, Krylov Anna I, Møller Klaus B, Coriani Sonia
DTU Chemistry, Technical University of Denmark, Kemitorvet Building 207, DK-2800 Kgs. Lyngby, Denmark.
Department of Chemistry, University of Southern California, Los Angeles, California 90089, USA.
Struct Dyn. 2021 Mar 12;8(2):024101. doi: 10.1063/4.0000070. eCollection 2021 Mar.
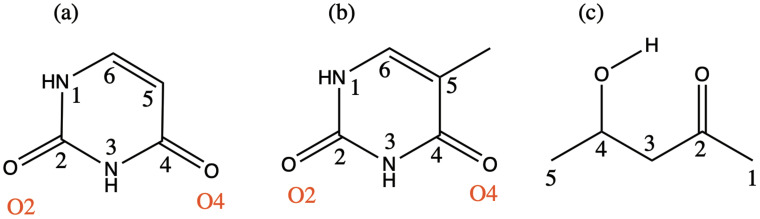
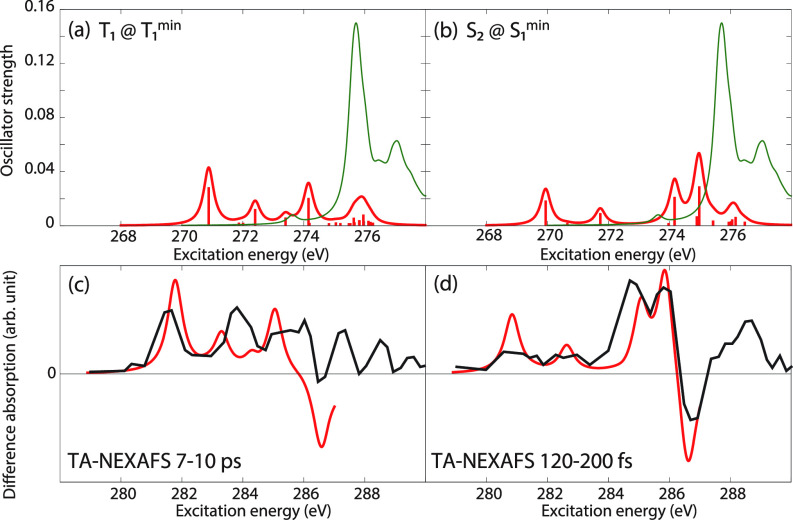
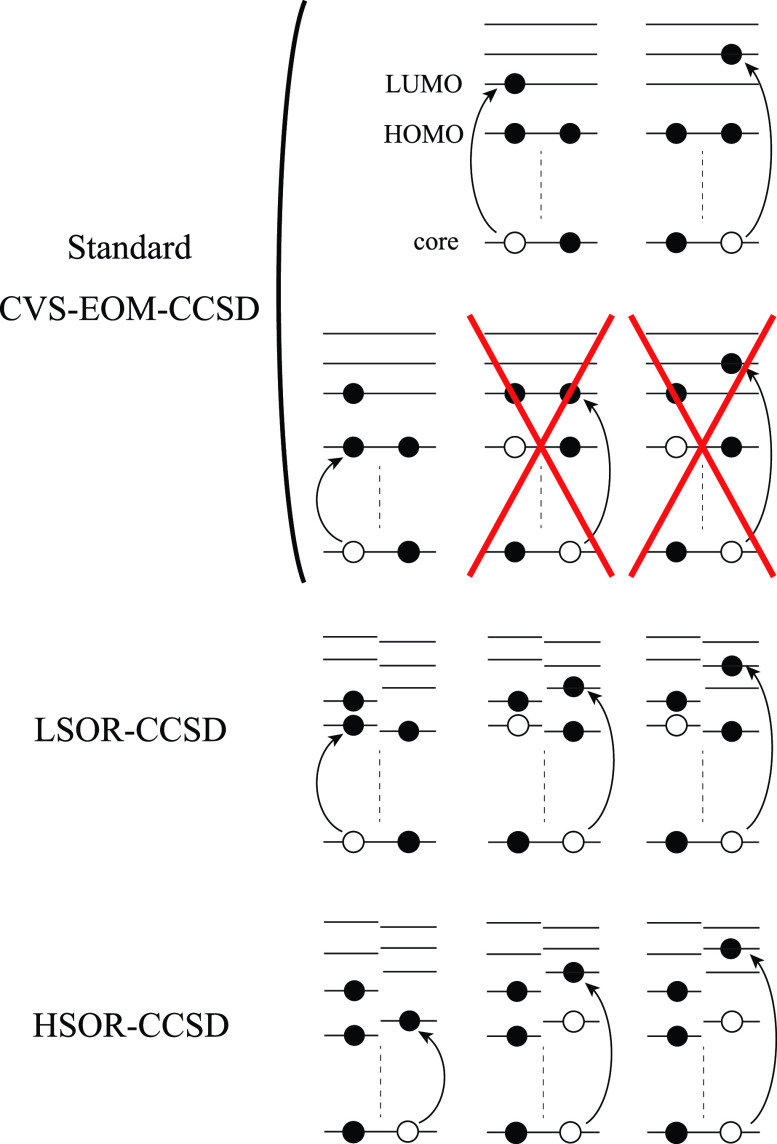
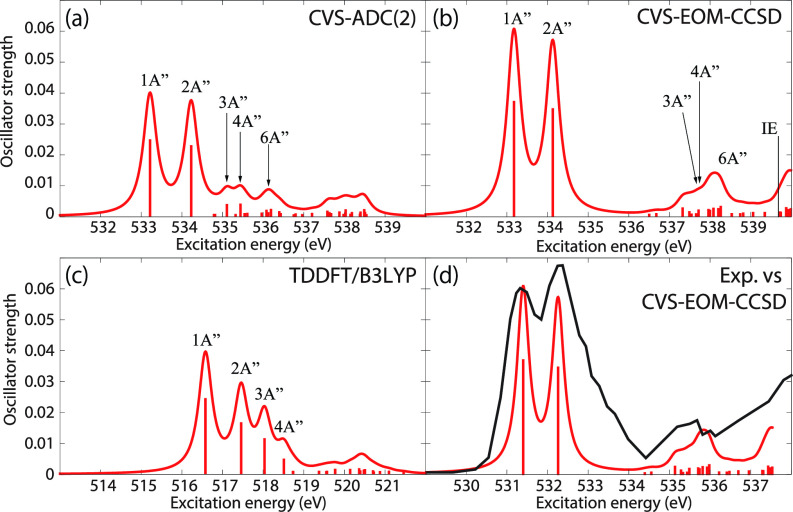
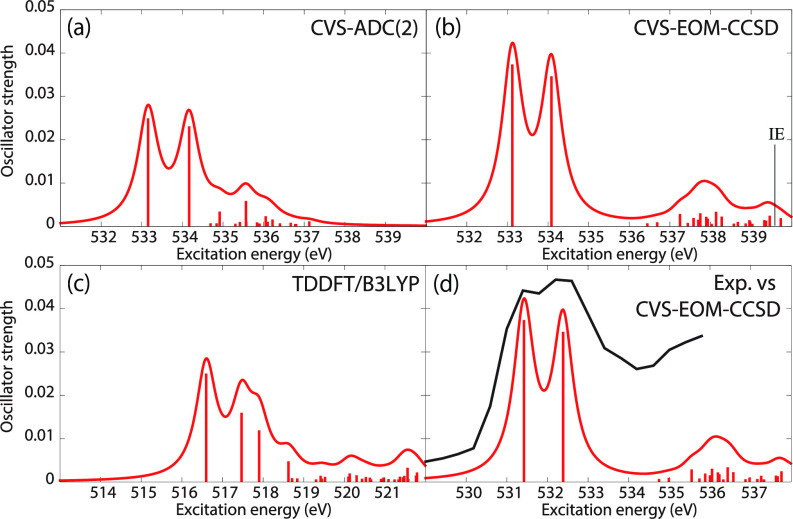
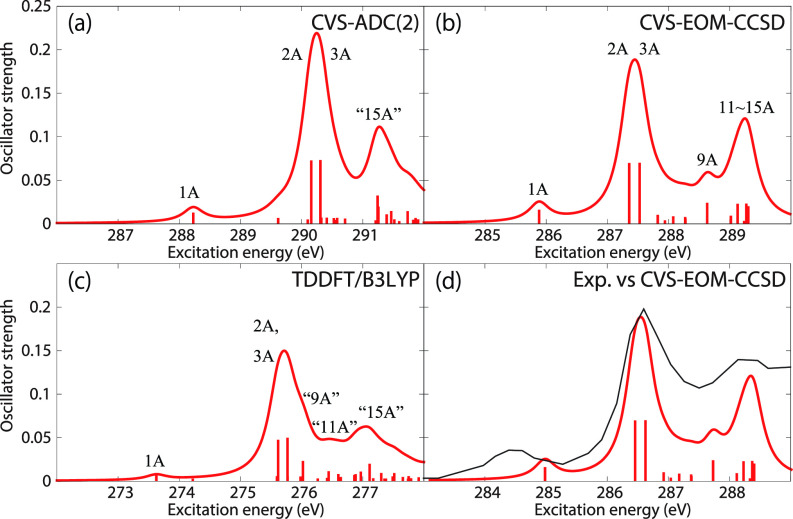
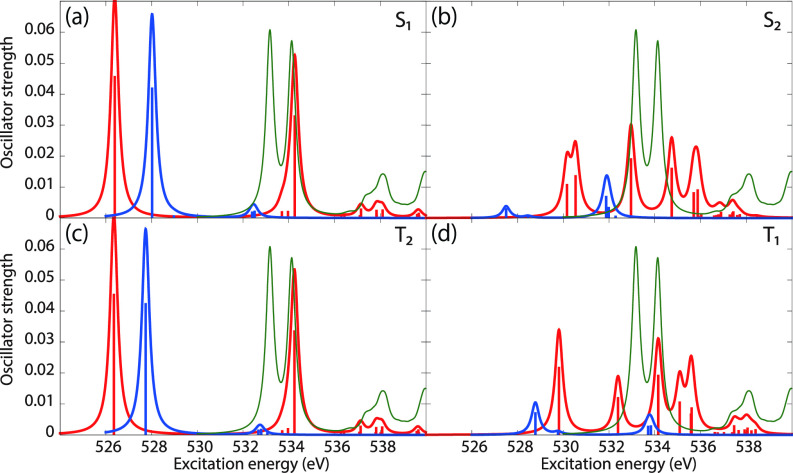
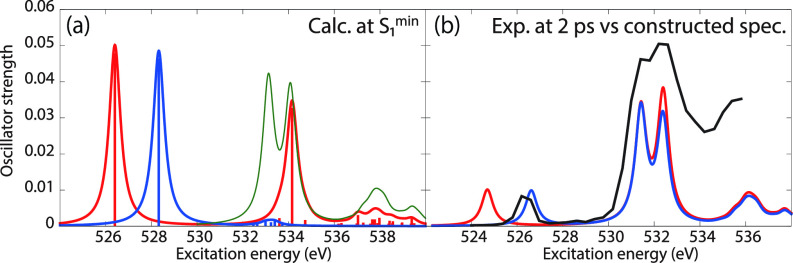
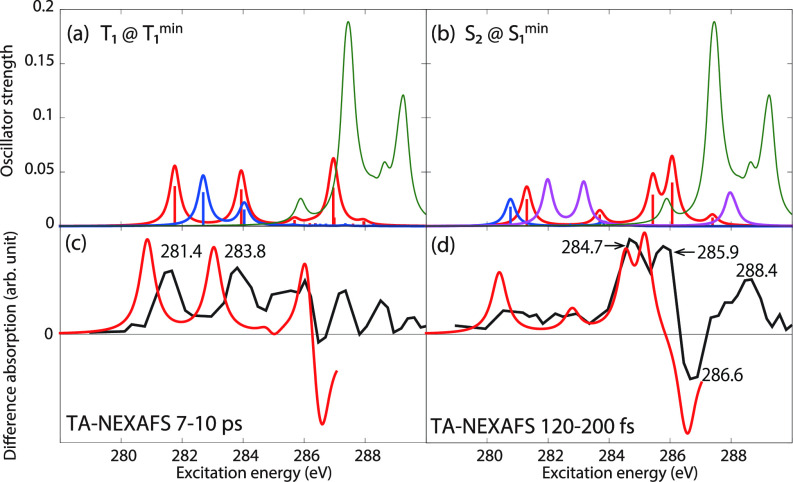
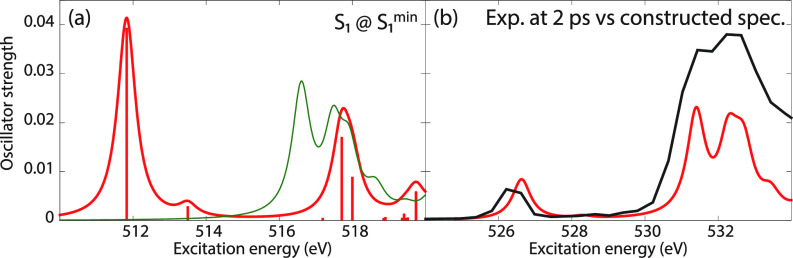
We assess the performance of different protocols for simulating excited-state x-ray absorption spectra. We consider three different protocols based on equation-of-motion coupled-cluster singles and doubles, two of them combined with the maximum overlap method. The three protocols differ in the choice of a reference configuration used to compute target states. Maximum-overlap-method time-dependent density functional theory is also considered. The performance of the different approaches is illustrated using uracil, thymine, and acetylacetone as benchmark systems. The results provide guidance for selecting an electronic structure method for modeling time-resolved x-ray absorption spectroscopy.
我们评估了用于模拟激发态X射线吸收光谱的不同协议的性能。我们考虑了基于运动方程耦合簇单双激发的三种不同协议,其中两种与最大重叠方法相结合。这三种协议在用于计算目标态的参考构型选择上有所不同。还考虑了最大重叠方法含时密度泛函理论。使用尿嘧啶、胸腺嘧啶和乙酰丙酮作为基准系统说明了不同方法的性能。结果为选择用于时间分辨X射线吸收光谱建模的电子结构方法提供了指导。