Zheng An, Lamkin Michael, Zhao Hanqing, Wu Cynthia, Su Hao, Gymrek Melissa
Department of Computer Science and Engineering, University of California San Diego, La Jolla, CA USA.
Department of Bioengineering, University of California San Diego, La Jolla, CA USA.
Nat Mach Intell. 2021 Feb;3(2):172-180. doi: 10.1038/s42256-020-00282-y. Epub 2021 Jan 18.
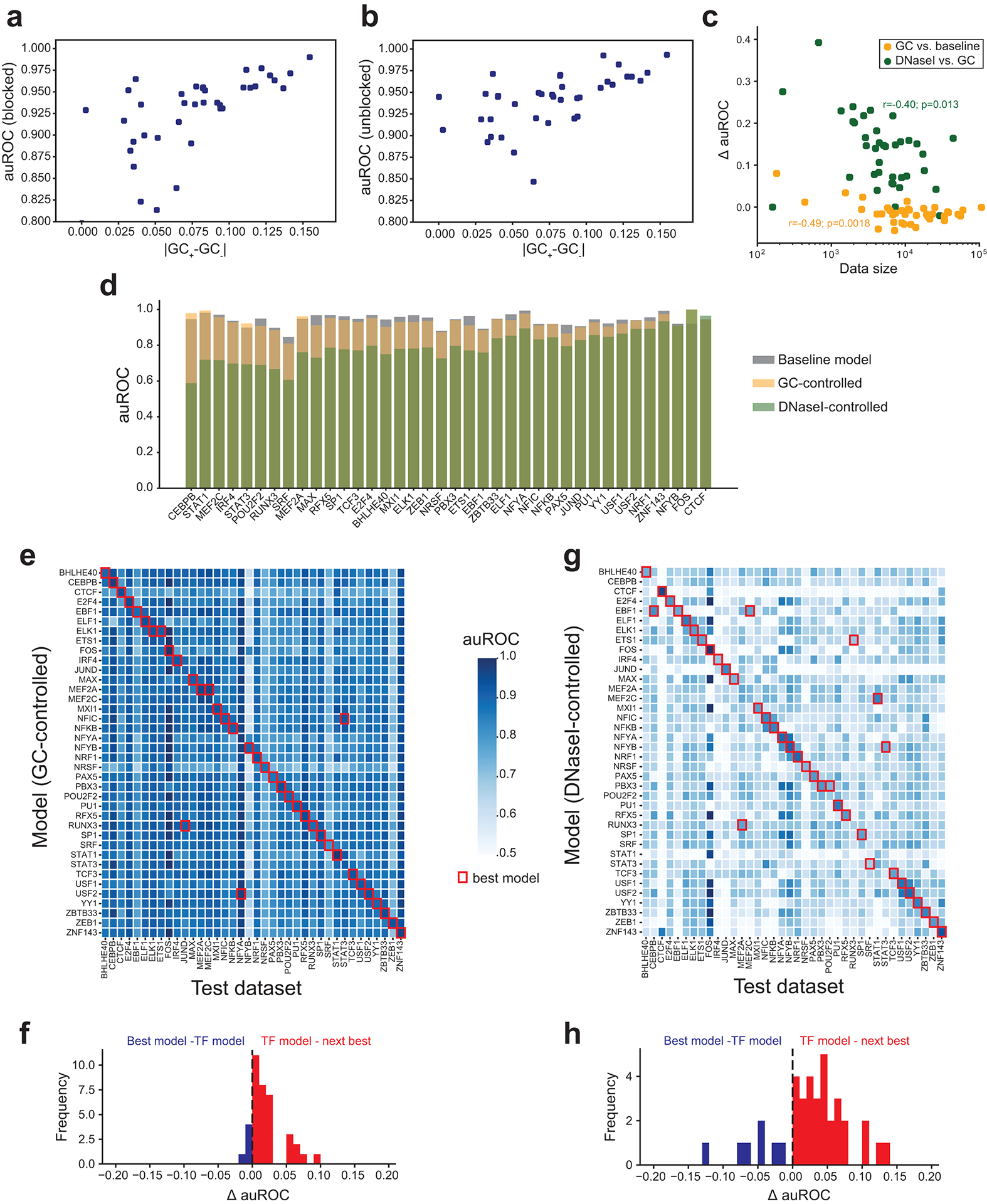
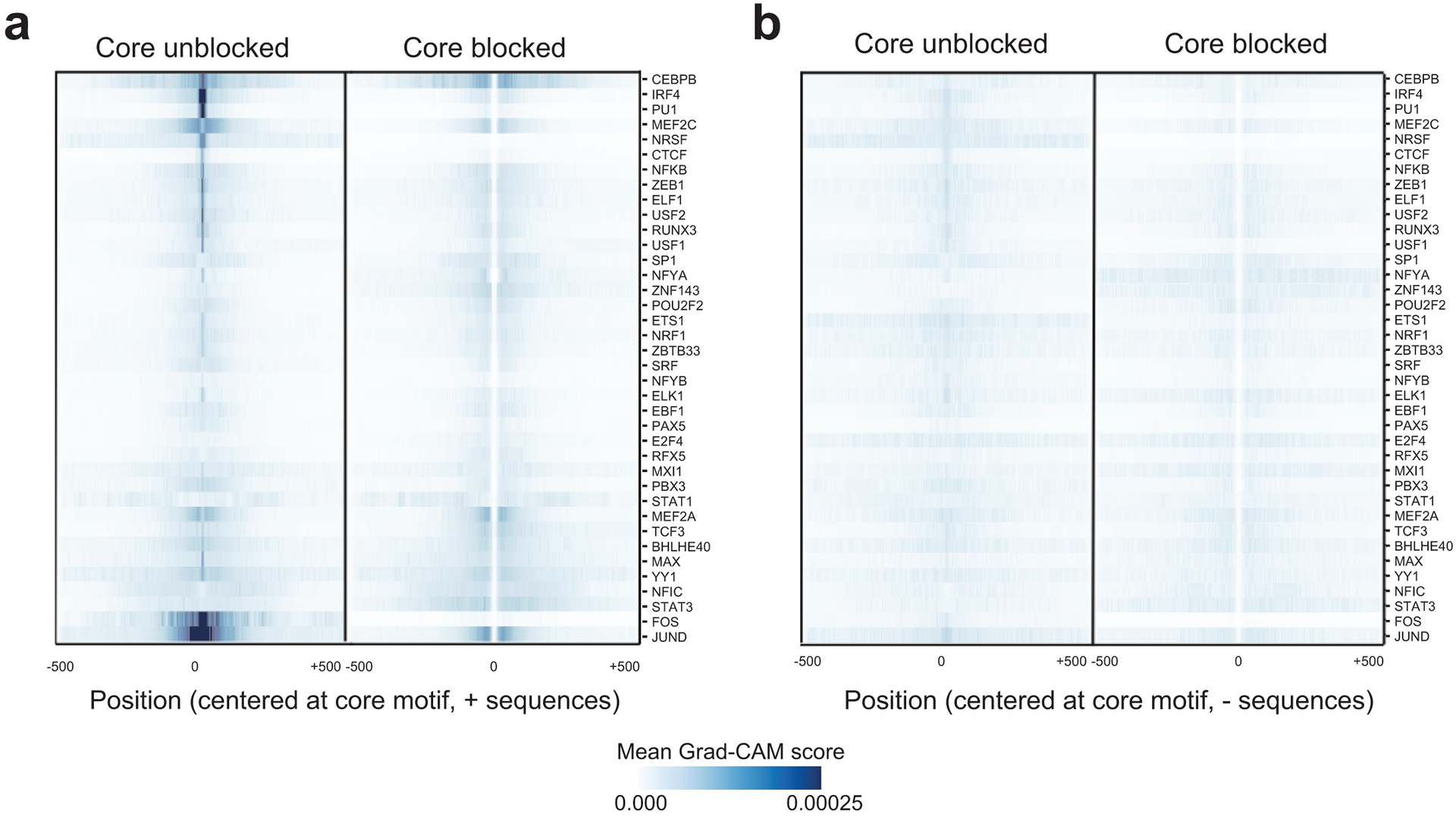
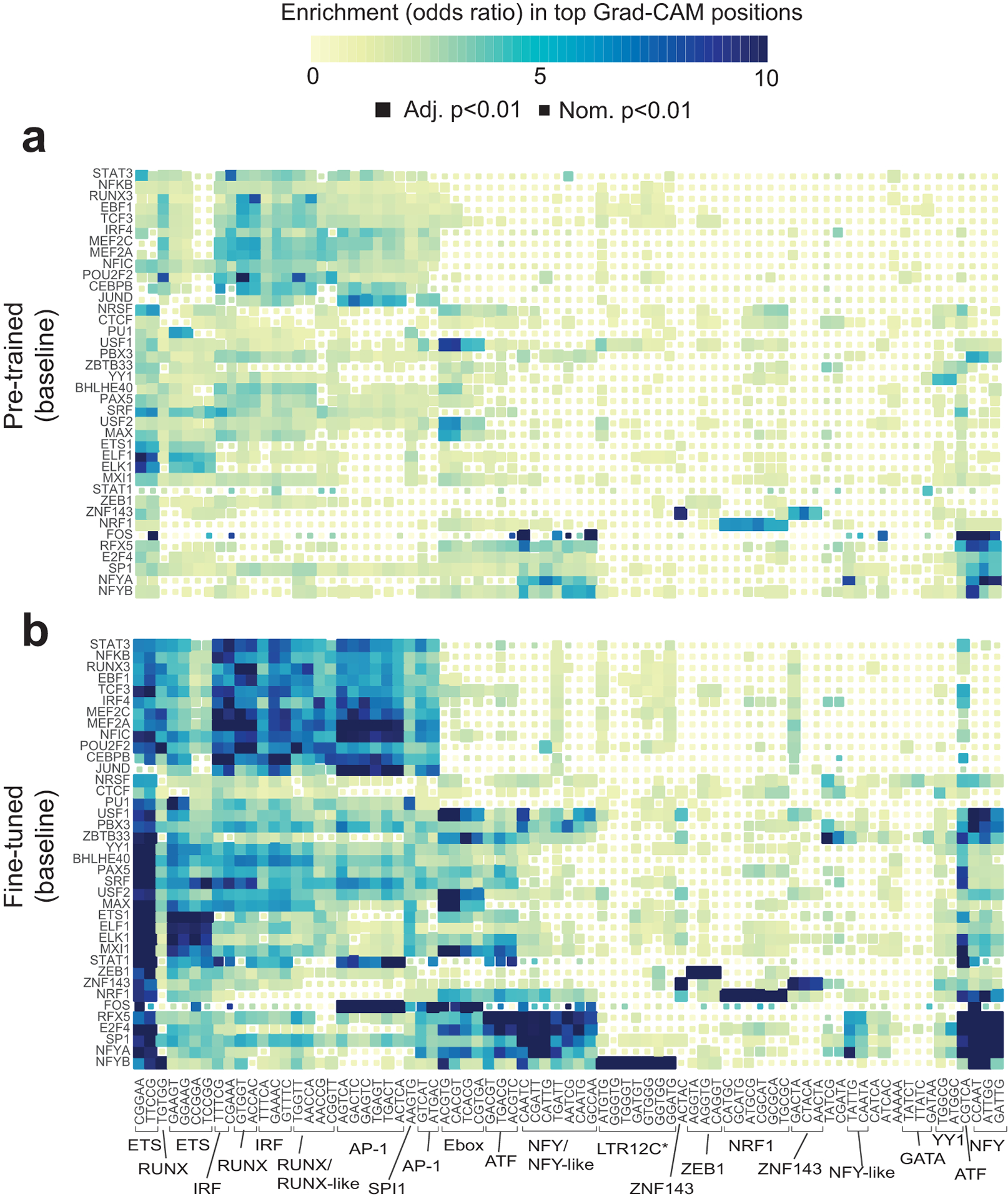
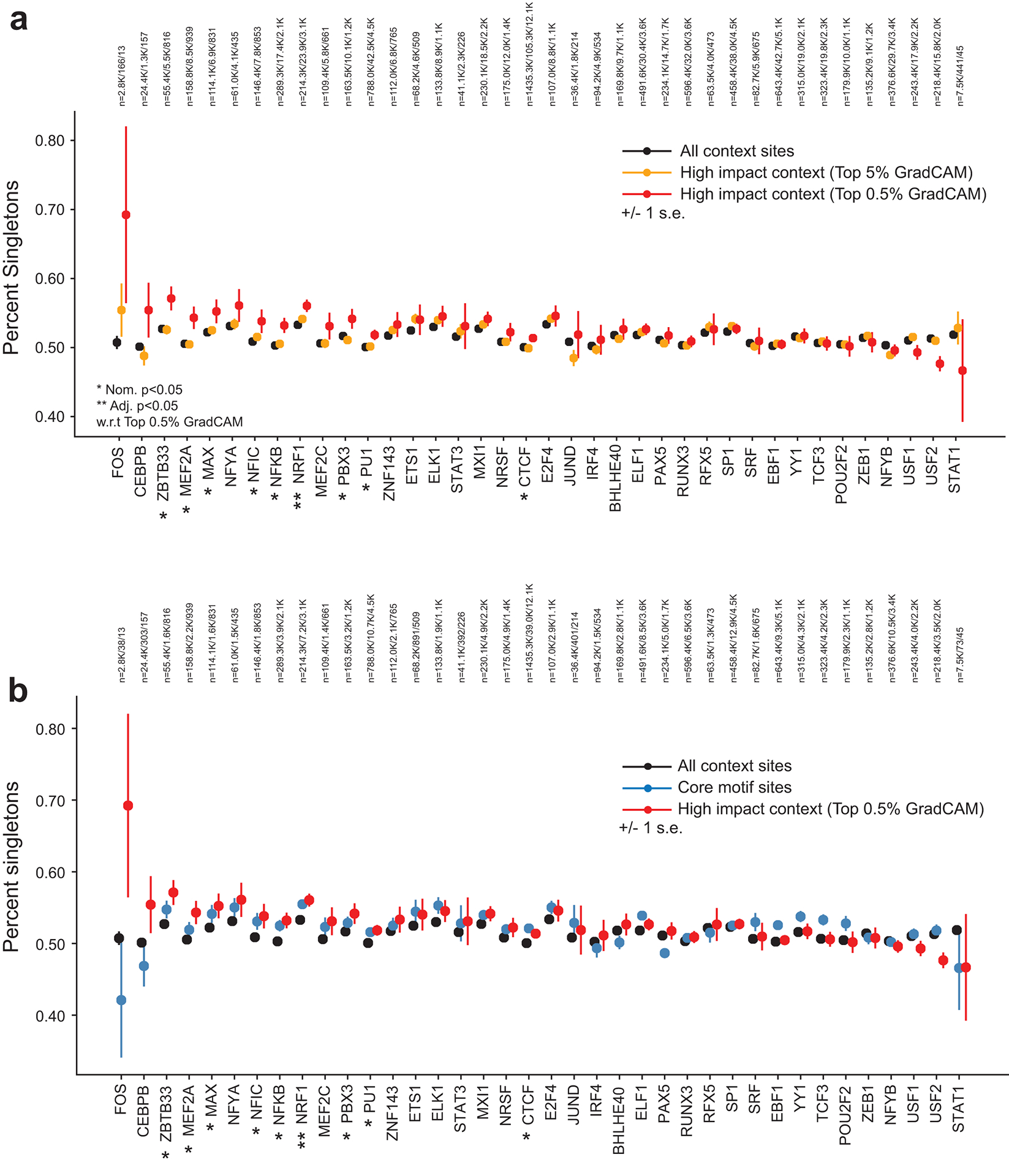
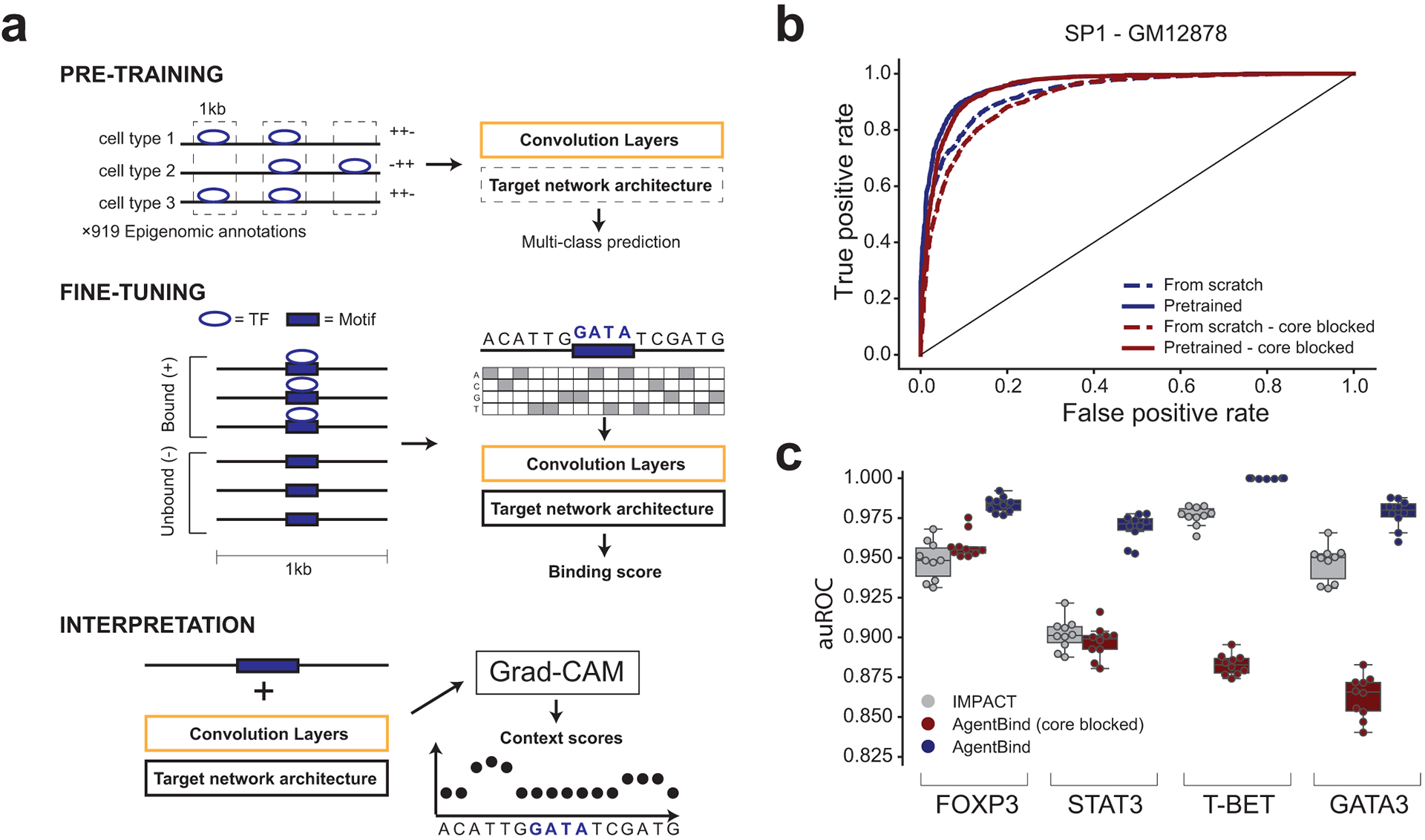
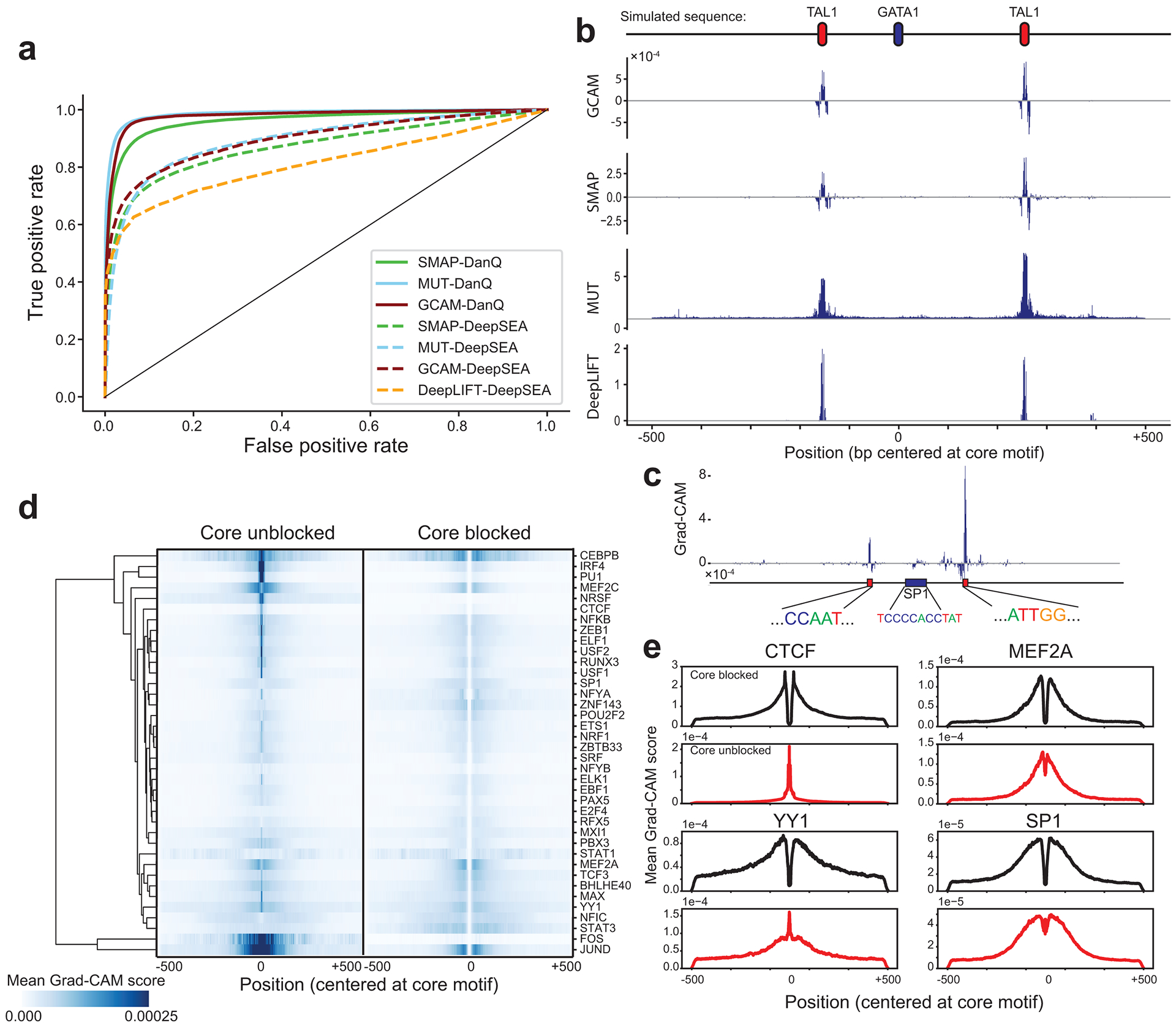
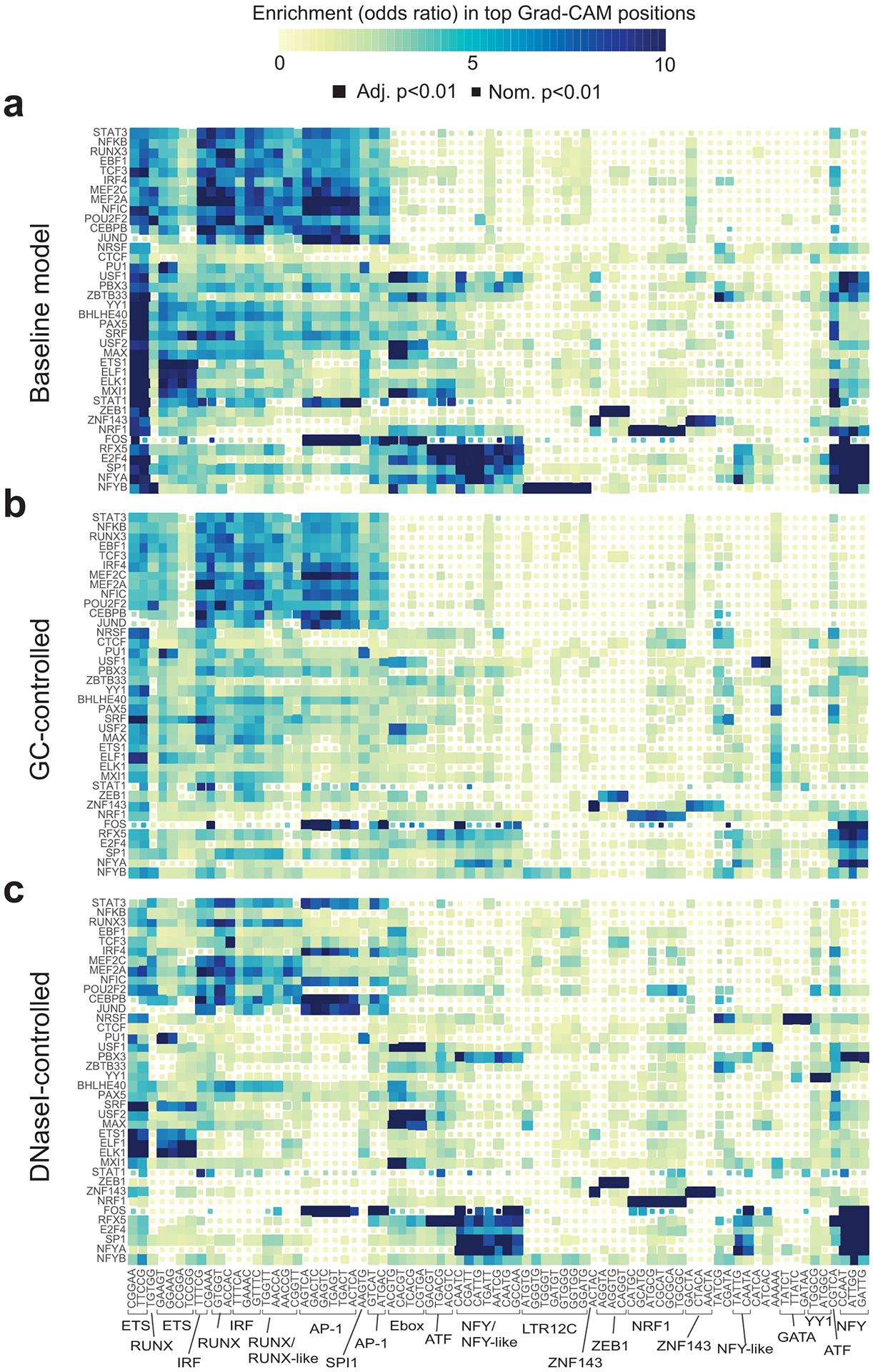
Transcription factors (TFs) bind DNA by recognizing specific sequence motifs, typically of length 6-12bp. A motif can occur many thousands of times in the human genome, but only a subset of those sites are actually bound. Here we present a machine learning framework leveraging existing convolutional neural network architectures and model interpretation techniques to identify and interpret sequence context features most important for predicting whether a particular motif instance will be bound. We apply our framework to predict binding at motifs for 38 TFs in a lymphoblastoid cell line, score the importance of context sequences at base-pair resolution, and characterize context features most predictive of binding. We find that the choice of training data heavily influences classification accuracy and the relative importance of features such as open chromatin. Overall, our framework enables novel insights into features predictive of TF binding and is likely to inform future deep learning applications to interpret non-coding genetic variants.
转录因子(TFs)通过识别特定的序列基序来结合DNA,这些基序通常长度为6 - 12个碱基对。一个基序在人类基因组中可能出现数千次,但实际上只有一部分位点会被结合。在这里,我们提出了一个机器学习框架,利用现有的卷积神经网络架构和模型解释技术,来识别和解释对于预测特定基序实例是否会被结合最为重要的序列上下文特征。我们应用我们的框架来预测淋巴母细胞系中38种转录因子基序的结合情况,以碱基对分辨率对上下文序列的重要性进行评分,并表征最能预测结合的上下文特征。我们发现训练数据的选择对分类准确性和诸如开放染色质等特征的相对重要性有很大影响。总体而言,我们的框架能够对预测转录因子结合的特征提供新的见解,并可能为未来解释非编码基因变异的深度学习应用提供参考。