Center for Data Sciences, Harvard Medical School, Boston, MA 02115, USA; Divisions of Genetics and Rheumatology, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA; Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA; Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA; Graduate School of Arts and Sciences, Harvard University, Cambridge, MA 02138, USA.
Center for Data Sciences, Harvard Medical School, Boston, MA 02115, USA; Divisions of Genetics and Rheumatology, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA; Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Am J Hum Genet. 2019 May 2;104(5):879-895. doi: 10.1016/j.ajhg.2019.03.012. Epub 2019 Apr 18.
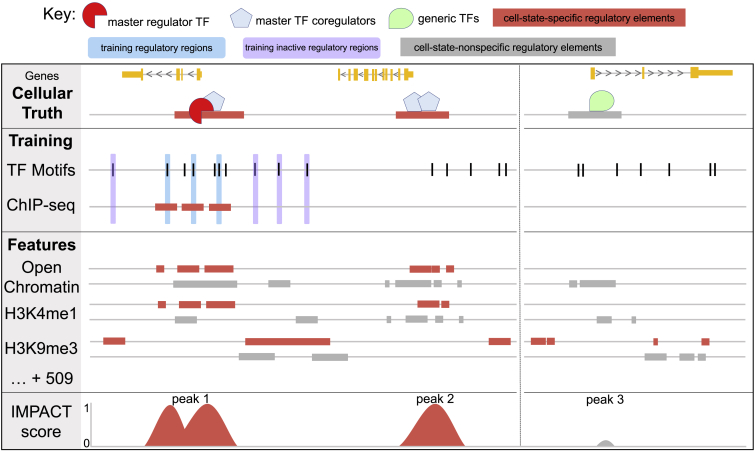
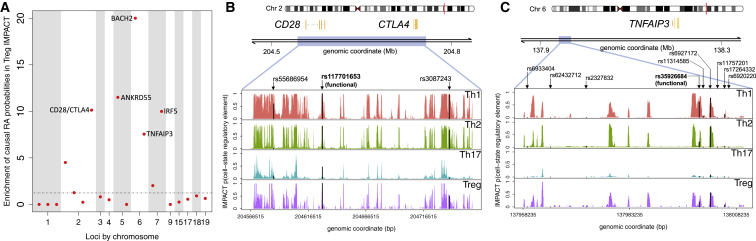
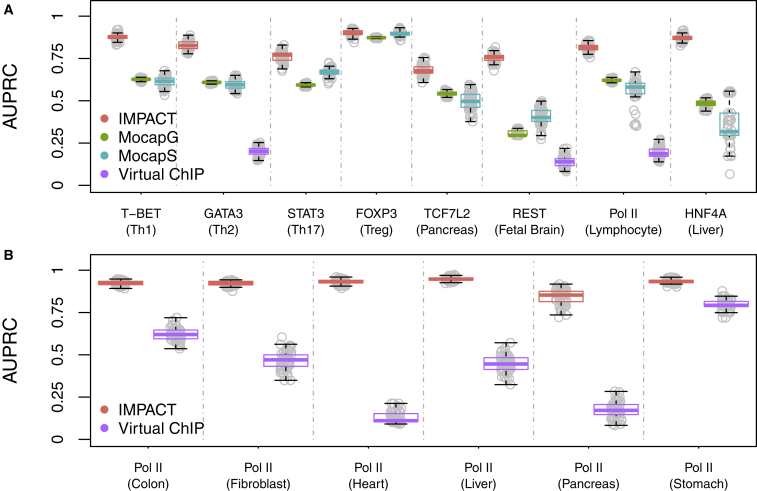
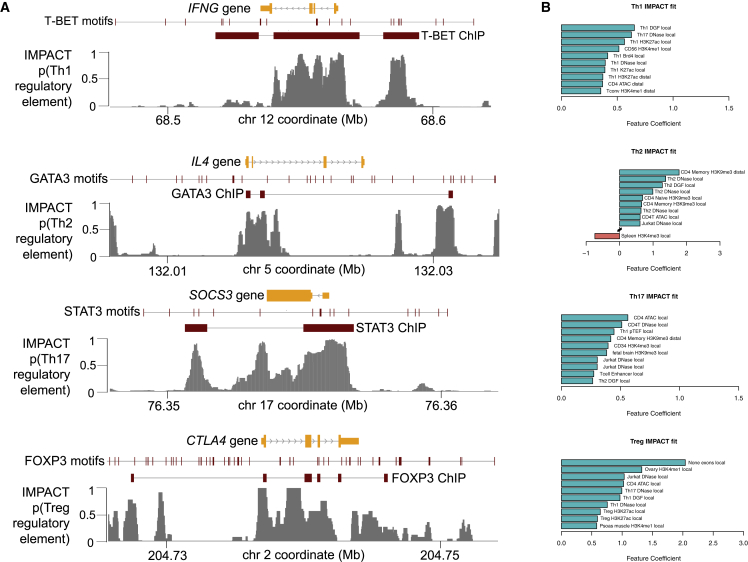
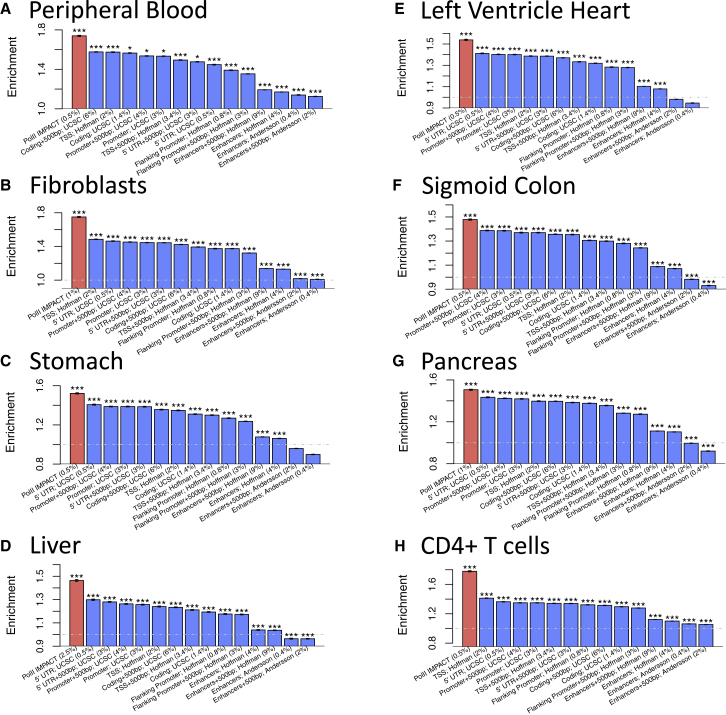
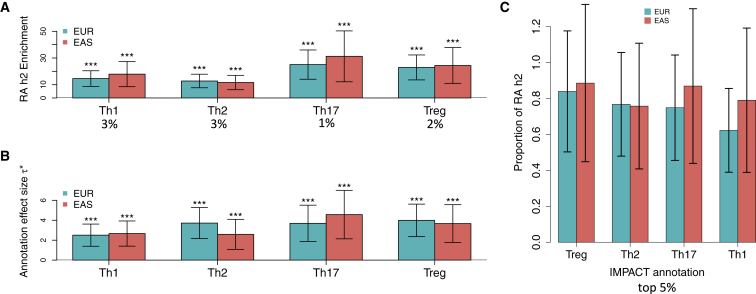
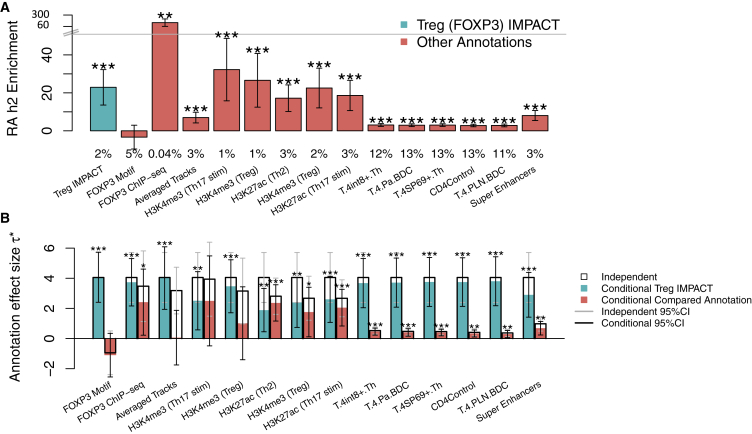
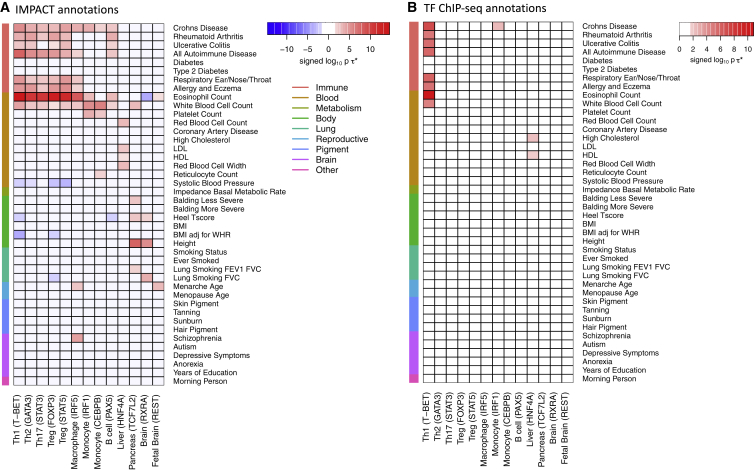
Despite significant progress in annotating the genome with experimental methods, much of the regulatory noncoding genome remains poorly defined. Here we assert that regulatory elements may be characterized by leveraging local epigenomic signatures where specific transcription factors (TFs) are bound. To link these two features, we introduce IMPACT, a genome annotation strategy that identifies regulatory elements defined by cell-state-specific TF binding profiles, learned from 515 chromatin and sequence annotations. We validate IMPACT using multiple compelling applications. First, IMPACT distinguishes between bound and unbound TF motif sites with high accuracy (average AUPRC 0.81, SE 0.07; across 8 tested TFs) and outperforms state-of-the-art TF binding prediction methods, MocapG, MocapS, and Virtual ChIP-seq. Second, in eight tested cell types, RNA polymerase II IMPACT annotations capture more cis-eQTL variation than sequence-based annotations, such as promoters and TSS windows (25% average increase in enrichment). Third, integration with rheumatoid arthritis (RA) summary statistics from European (N = 38,242) and East Asian (N = 22,515) populations revealed that the top 5% of CD4 Treg IMPACT regulatory elements capture 85.7% of RA h2, the most comprehensive explanation for RA h2 to date. In comparison, the average RA h2 captured by compared CD4 T histone marks is 42.3% and by CD4 T specifically expressed gene sets is 36.4%. Lastly, we find that IMPACT may be used in many different cell types to identify complex trait associated regulatory elements.
尽管在使用实验方法对基因组进行注释方面取得了重大进展,但大部分调控性非编码基因组仍未得到很好的定义。在这里,我们断言,通过利用特定转录因子(TF)结合的局部表观基因组特征,可以对调控元件进行特征描述。为了将这两个特征联系起来,我们引入了 IMPACT,这是一种基因组注释策略,它可以识别由细胞状态特异性 TF 结合谱定义的调控元件,这些谱是从 515 个染色质和序列注释中学习到的。我们使用多种引人注目的应用程序来验证 IMPACT。首先,IMPACT 可以以高精度(平均 AUPRC 为 0.81,SE 为 0.07;在 8 个测试的 TF 中)区分结合和未结合的 TF 基序位点,并且优于最先进的 TF 结合预测方法 MocapG、MocapS 和 Virtual ChIP-seq。其次,在八个测试的细胞类型中,RNA 聚合酶 II 的 IMPACT 注释比基于序列的注释(例如启动子和 TSS 窗口)捕获更多的顺式-eQTL 变异(平均富集度增加 25%)。第三,与来自欧洲(N = 38,242)和东亚(N = 22,515)人群的类风湿关节炎(RA)汇总统计数据的整合表明,TOP5%的 CD4 Treg IMPACT 调控元件捕获了 RA h2 的 85.7%,这是迄今为止对 RA h2 最全面的解释。相比之下,比较 CD4 T 组蛋白标记平均捕获的 RA h2 为 42.3%,比较 CD4 T 特异性表达基因集捕获的 RA h2 为 36.4%。最后,我们发现,IMPACT 可以在许多不同的细胞类型中用于识别与复杂性状相关的调控元件。