Gastroenterology and Digestive Endoscopy Unit, IRCCS Policlinico San Donato, 20097 Milano, Italy.
Neuromuscular Diseases and Neuroimmunology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milano, Italy.
Int J Mol Sci. 2021 Mar 11;22(6):2850. doi: 10.3390/ijms22062850.
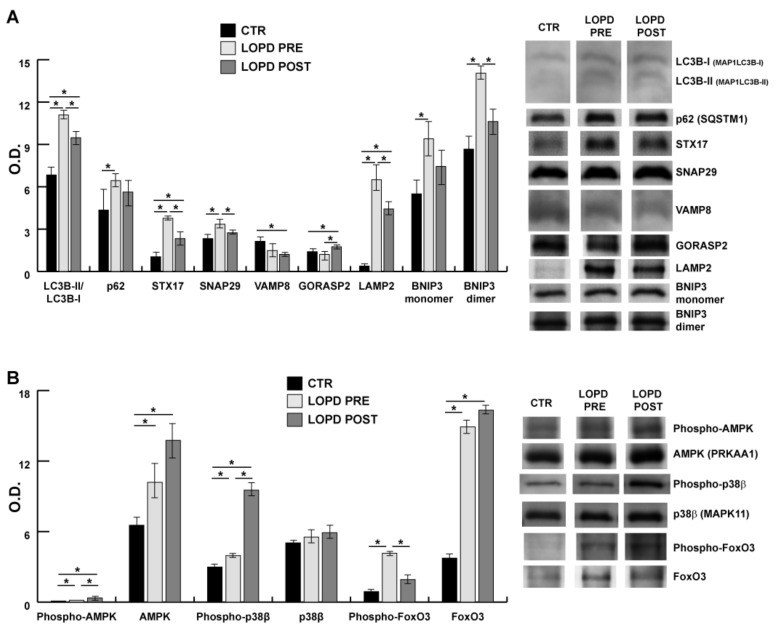
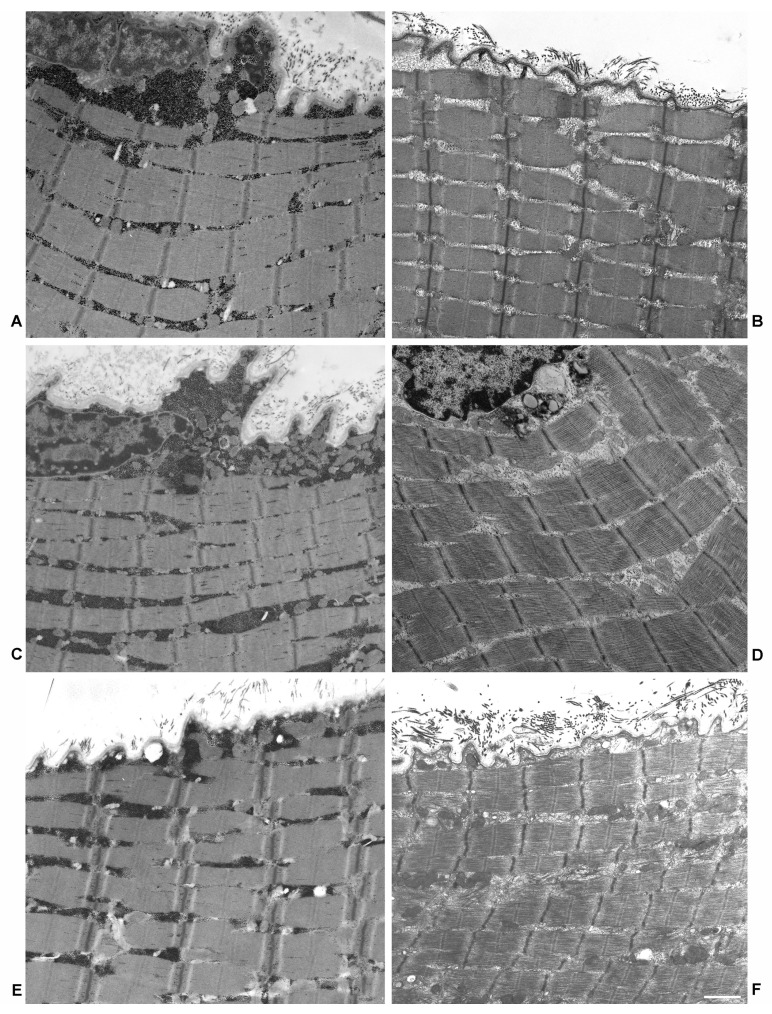
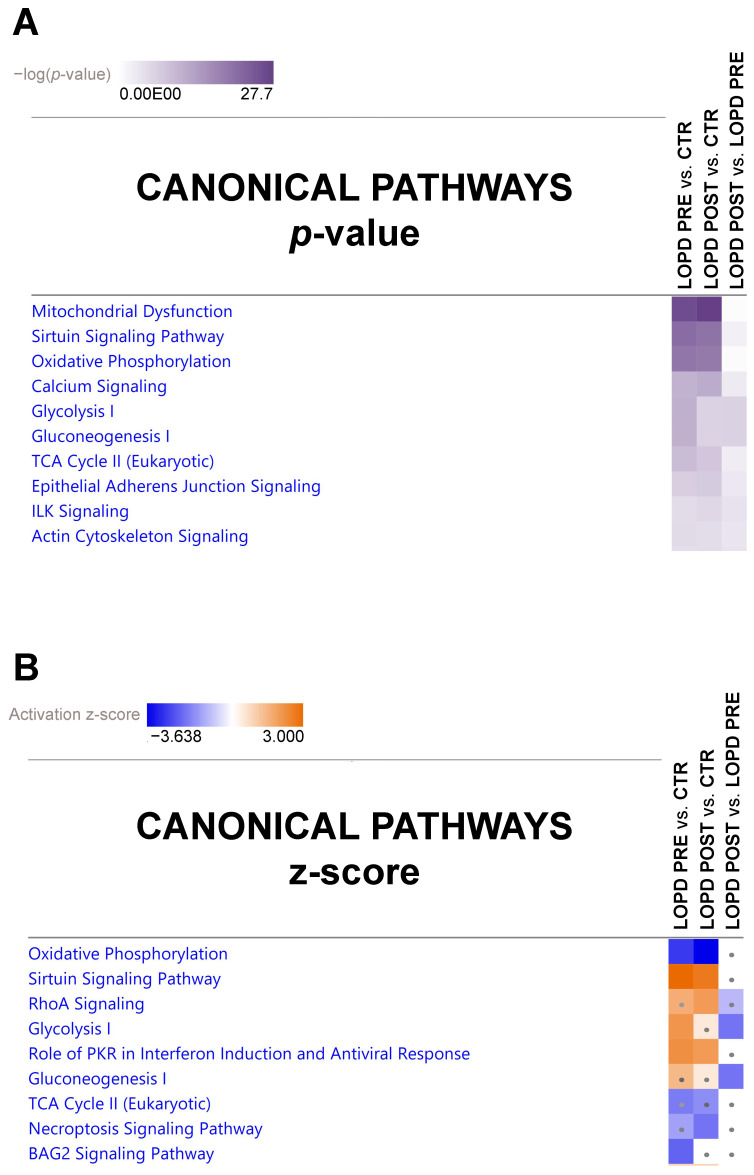
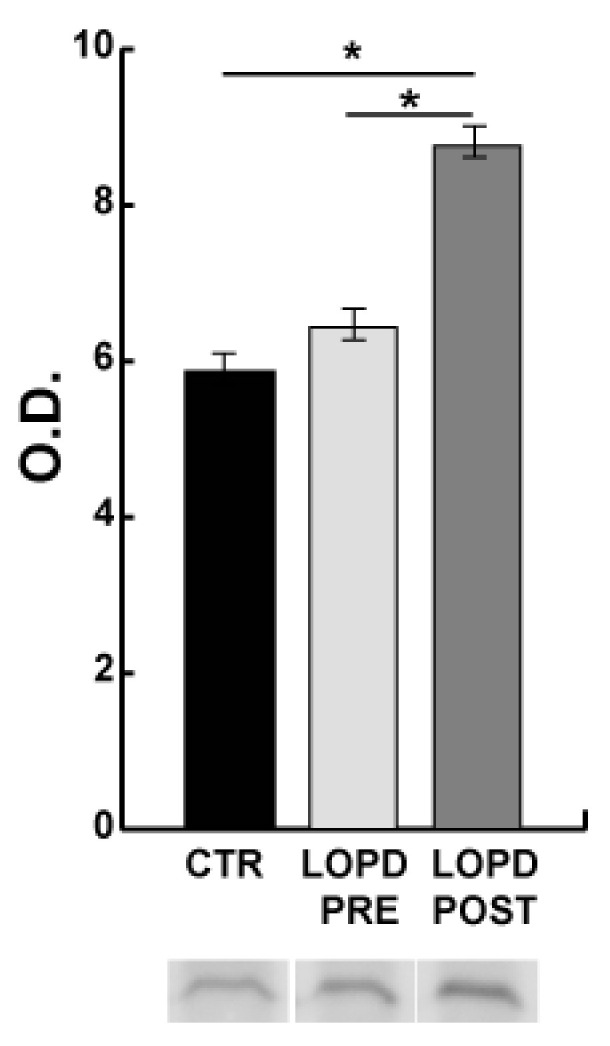
Mutations in the acidic alpha-glucosidase (GAA) coding gene cause Pompe disease. Late-onset Pompe disease (LOPD) is characterized by progressive proximal and axial muscle weakness and atrophy, causing respiratory failure. Enzyme replacement therapy (ERT), based on recombinant human GAA infusions, is the only available treatment; however, the efficacy of ERT is variable. Here we address the question whether proteins at variance in LOPD muscle of patients before and after 1 year of ERT, compared withhealthy age-matched subjects (CTR), reveal a specific signature. Proteins extracted from skeletal muscle of LOPD patients and CTR were analyzed by combining gel based (two-dimensional difference gel electrophoresis) and label-free (liquid chromatography-mass spectrometry) proteomic approaches, and ingenuity pathway analysis. Upstream regulators targeting autophagy and lysosomal tethering were assessed by immunoblotting. 178 proteins were changed in abundance in LOPD patients, 47 of them recovered normal level after ERT. Defects in oxidative metabolism, muscle contractile protein regulation, cytoskeletal rearrangement, and membrane reorganization persisted. Metabolic changes, ER stress and UPR (unfolded protein response) contribute to muscle proteostasis dysregulation with active membrane remodeling (high levels of LC3BII/LC3BI) and accumulation of p62, suggesting imbalance in the autophagic process. Active lysosome biogenesis characterizes both LOPD PRE and POST, unparalleled by molecules involved in lysosome tethering (VAMP8, SNAP29, STX17, and GORASP2) and BNIP3. In conclusion this study reveals a specific signature that suggests ERT prolongation and molecular targets to ameliorate patient's outcome.
酸性α-葡萄糖苷酶(GAA)编码基因突变导致庞贝病。迟发性庞贝病(LOPD)的特征是进行性近端和轴性肌肉无力和萎缩,导致呼吸衰竭。基于重组人 GAA 输注的酶替代疗法(ERT)是唯一可用的治疗方法;然而,ERT 的疗效是可变的。在这里,我们探讨了一个问题,即在 ERT 治疗 1 年后的 LOPD 患者肌肉中,与健康年龄匹配的对照组(CTR)相比,蛋白质是否存在差异,从而揭示出一种特定的特征。通过结合凝胶基础(二维差异凝胶电泳)和无标记(液相色谱-质谱)蛋白质组学方法以及 ingenuity 通路分析,分析了从 LOPD 患者和 CTR 提取的骨骼肌中的蛋白质。通过免疫印迹评估了针对自噬和溶酶体连接的上游调节剂。在 LOPD 患者中,有 178 种蛋白质的丰度发生了变化,其中 47 种在 ERT 后恢复正常水平。氧化代谢、肌肉收缩蛋白调节、细胞骨架重排和膜重组的缺陷仍然存在。代谢变化、内质网应激和 UPR(未折叠蛋白反应)导致肌肉蛋白稳态失调,伴有活跃的膜重塑(LC3BII/LC3BI 水平升高)和 p62 的积累,表明自噬过程失衡。溶酶体生物发生的活跃特征在 LOPD PRE 和 POST 中都存在,这是前所未有的,与溶酶体连接(VAMP8、SNAP29、STX17 和 GORASP2)和 BNIP3 涉及的分子不同。总之,这项研究揭示了一种特定的特征,提示 ERT 延长和分子靶标可改善患者的预后。