Department of Medical Genetics, Medical University, Pawinskiego 3c, 02-106 Warsaw, Poland.
Mossakowski Medical Research Center, Department of Experimental and Clinical Neuropathology, Polish Academy of Sciences, 02-106 Warsaw, Poland.
Genes (Basel). 2021 Mar 25;12(4):468. doi: 10.3390/genes12040468.
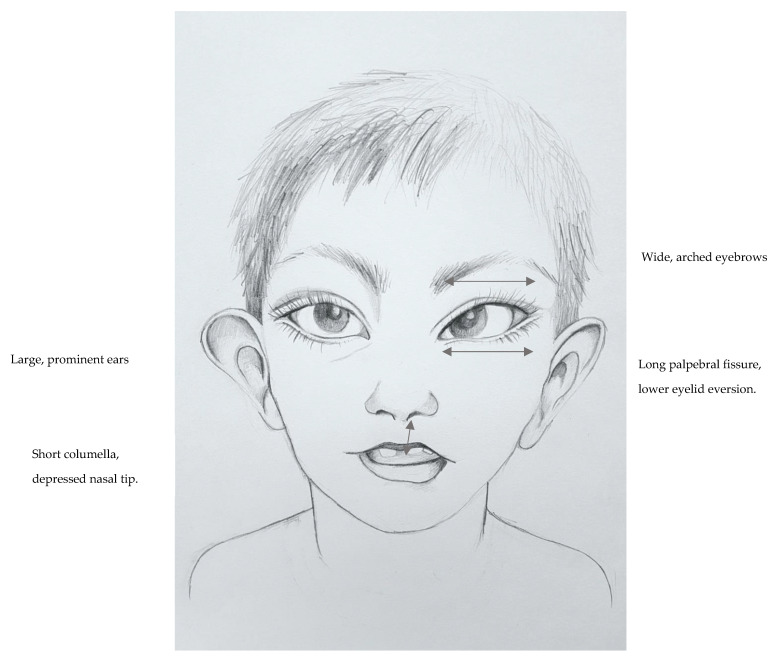
Kabuki syndrome (KS) is a rare developmental disorder principally comprised of developmental delay, hypotonia and a clearly defined dysmorphism: elongation of the structures surrounding the eyes, a shortened and depressed nose, thinning of the upper lip and thickening of the lower lip, large and prominent ears, hypertrichosis and scoliosis. Other characteristics include poor physical growth, cardiac, gastrointestinal and renal anomalies as well as variable behavioral issues, including autistic features. De novo or inherited pathogenic/likely pathogenic variants in the gene are the most common cause of KS and account for up to 75% of patients. Variants in cause up to 5% of cases (X-linked dominant inheritance), while the etiology of about 20% of cases remains unknown. Current KS diagnostic criteria include hypotonia during infancy, developmental delay and/or intellectual disability, typical dysmorphism and confirmed pathogenic/likely pathogenic variant in or . Care for KS patients includes the control of physical and psychomotor development during childhood, rehabilitation and multi-specialist care. This paper reviews the current clinical knowledge, provides molecular and scientific links and sheds light on the treatment of Kabuki syndrome individuals.
歌舞伎综合征(KS)是一种罕见的发育障碍疾病,主要表现为发育迟缓、肌张力低下和明显的畸形:眼睛周围结构拉长、鼻梁短而凹陷、上唇变薄、下唇变厚、耳朵大而突出、多毛症和脊柱侧凸。其他特征包括生长不良、心脏、胃肠道和肾脏异常以及各种行为问题,包括自闭症特征。 基因中的新生或遗传致病性/可能致病性变异是 KS 最常见的原因,占患者的 75%。 基因中的变异占病例的 5%(X 连锁显性遗传),而约 20%的病例的病因仍不清楚。目前的 KS 诊断标准包括婴儿期的肌张力低下、发育迟缓及/或智力障碍、典型的畸形以及 或 中已确认的致病性/可能致病性变异。KS 患者的治疗包括儿童期身体和运动发育的控制、康复和多学科护理。本文综述了目前的临床知识,提供了分子和科学联系,并阐明了治疗歌舞伎综合征个体的方法。