Department of Cardiology, Youjiang Medical University for Nationalities, Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, 533000 Guangxi, China.
Department of Neurology, Youjiang Medical University for Nationalities, Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, 533000 Guangxi, China.
Biomed Res Int. 2021 Mar 25;2021:6644827. doi: 10.1155/2021/6644827. eCollection 2021.
This study is aimed at understanding the molecular mechanisms and exploring potential therapeutic targets for atrial fibrillation (AF) by multiomics analysis.
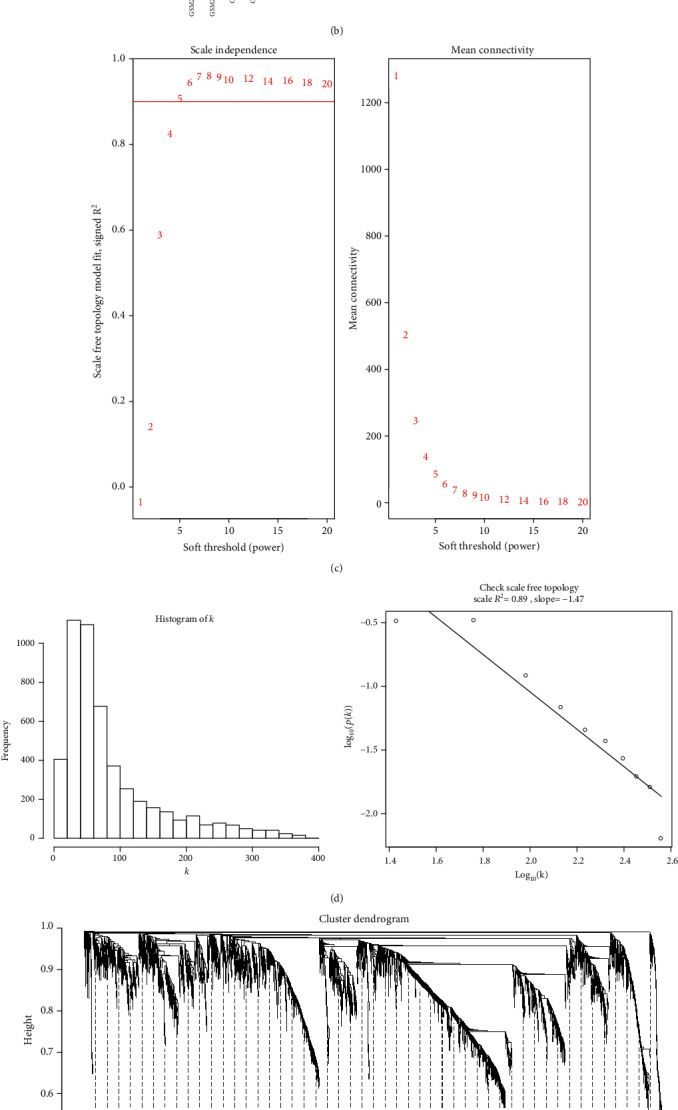
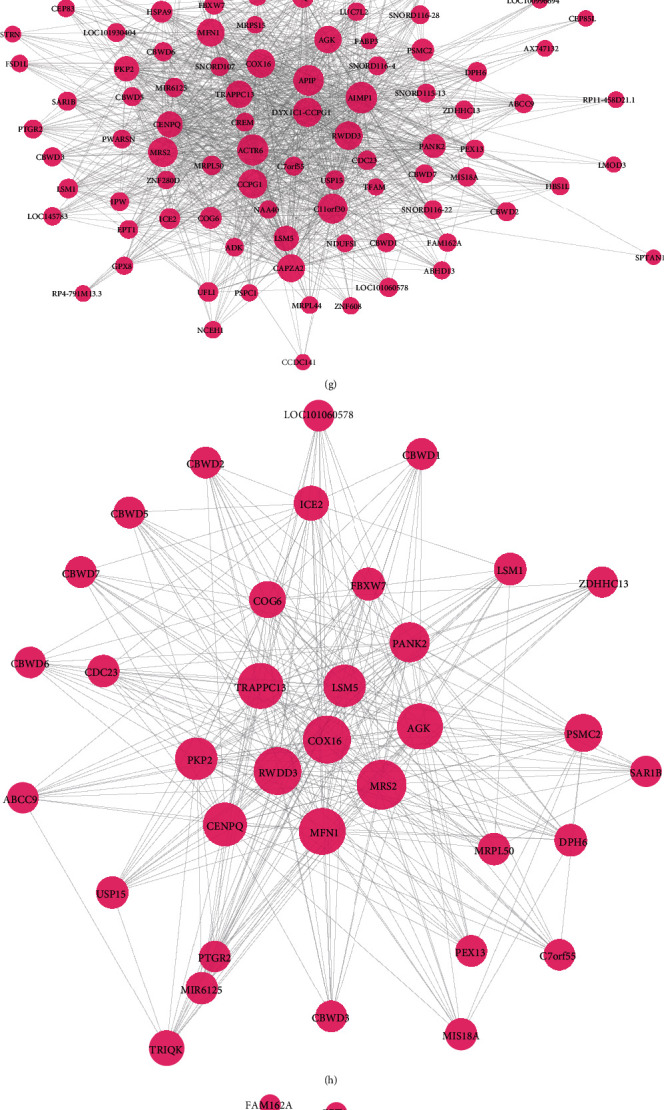
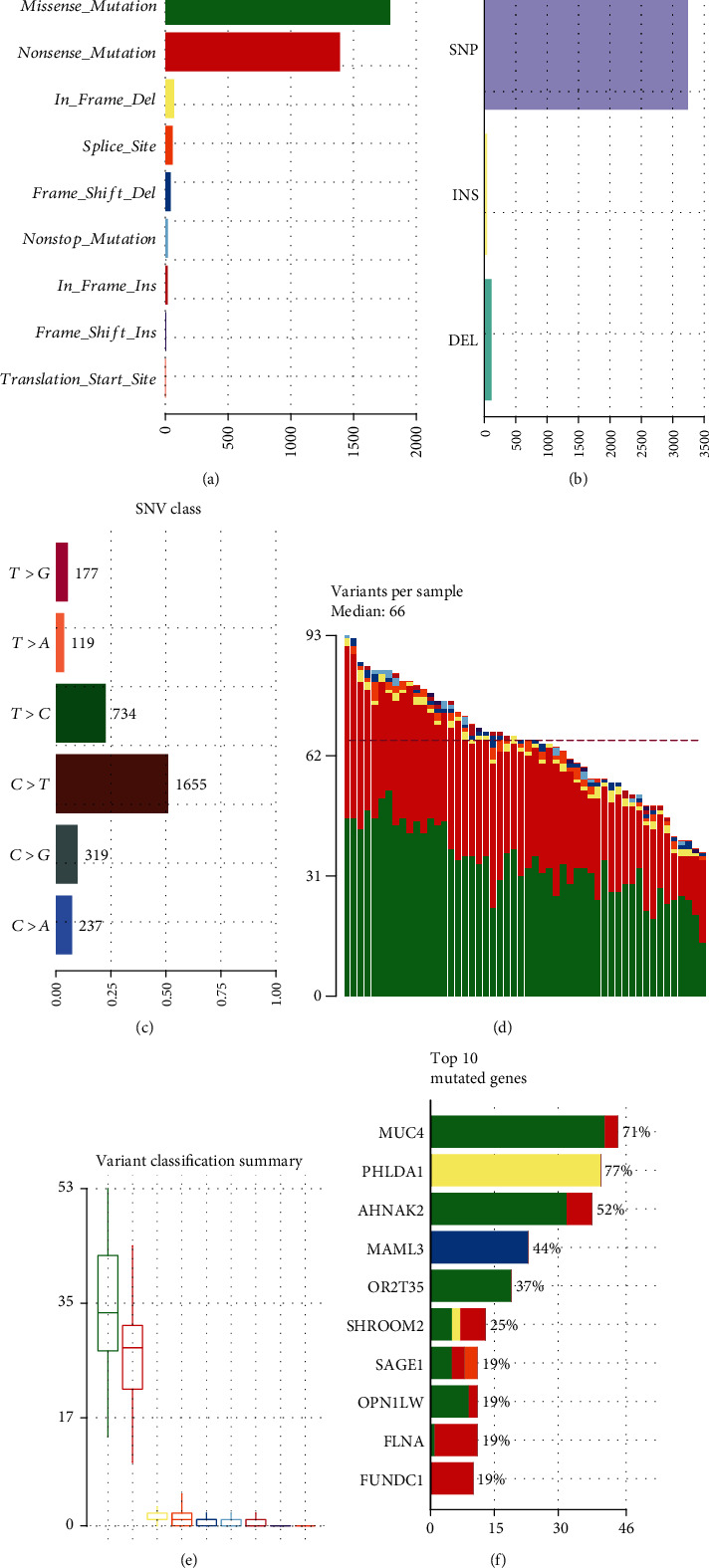
Transcriptomics and methylation data of AF patients were retrieved from the Gene Expression Omnibus (GEO). Differentially expressed genes (DEGs) and differentially methylated sites between AF and normal samples were screened. Then, highly expressed and hypomethylated and lowly expressed and hypermethylated genes were identified for AF. Weighted gene coexpression network analysis (WGCNA) was presented to construct AF-related coexpression networks. 52 AF blood samples were used for whole exome sequence. The mutation was visualized by the maftools package in R. Key genes were validated in AF using independent datasets.
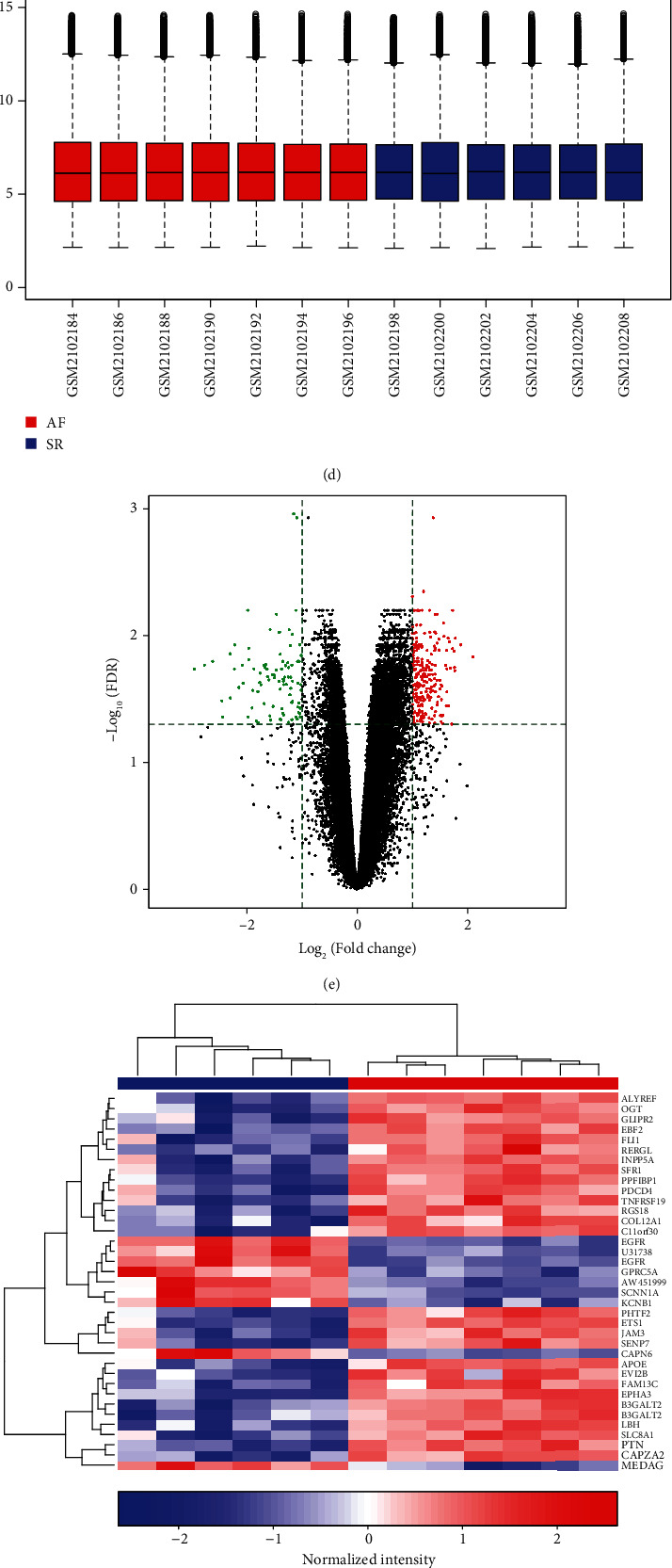
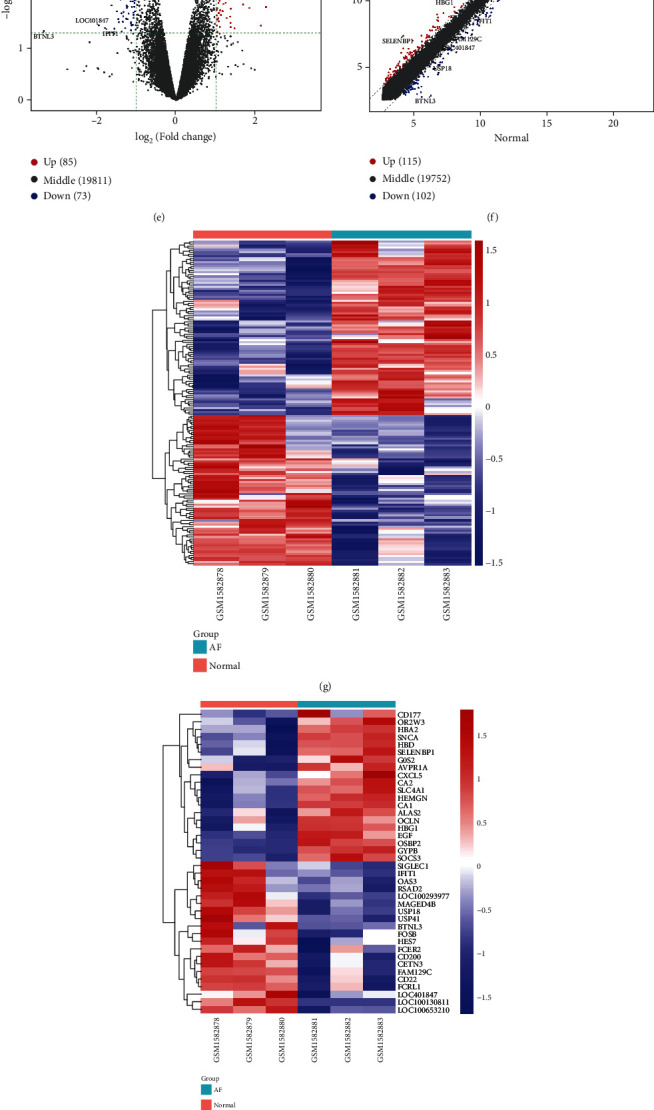
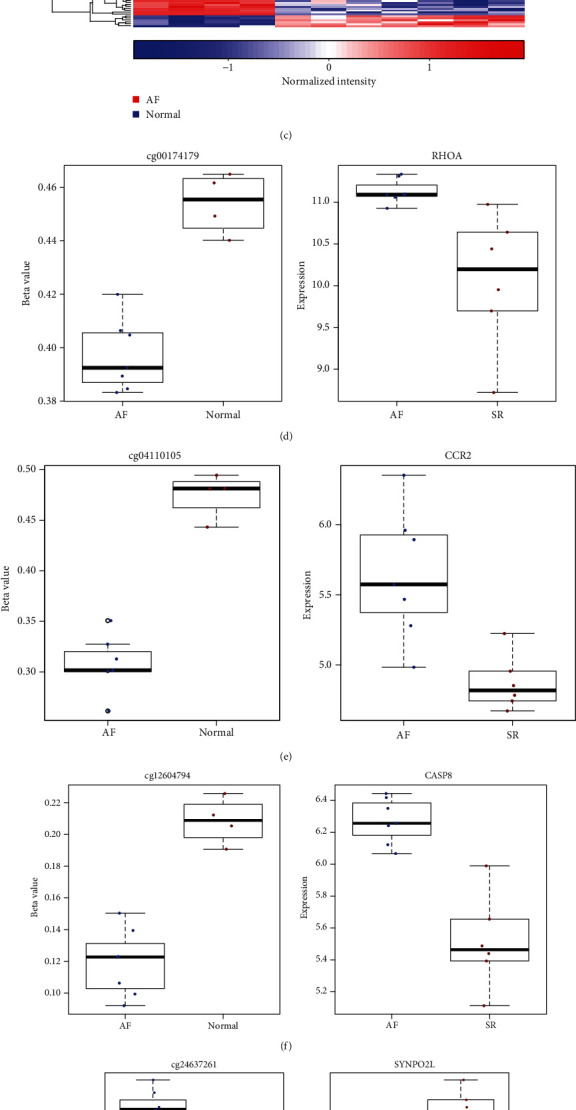
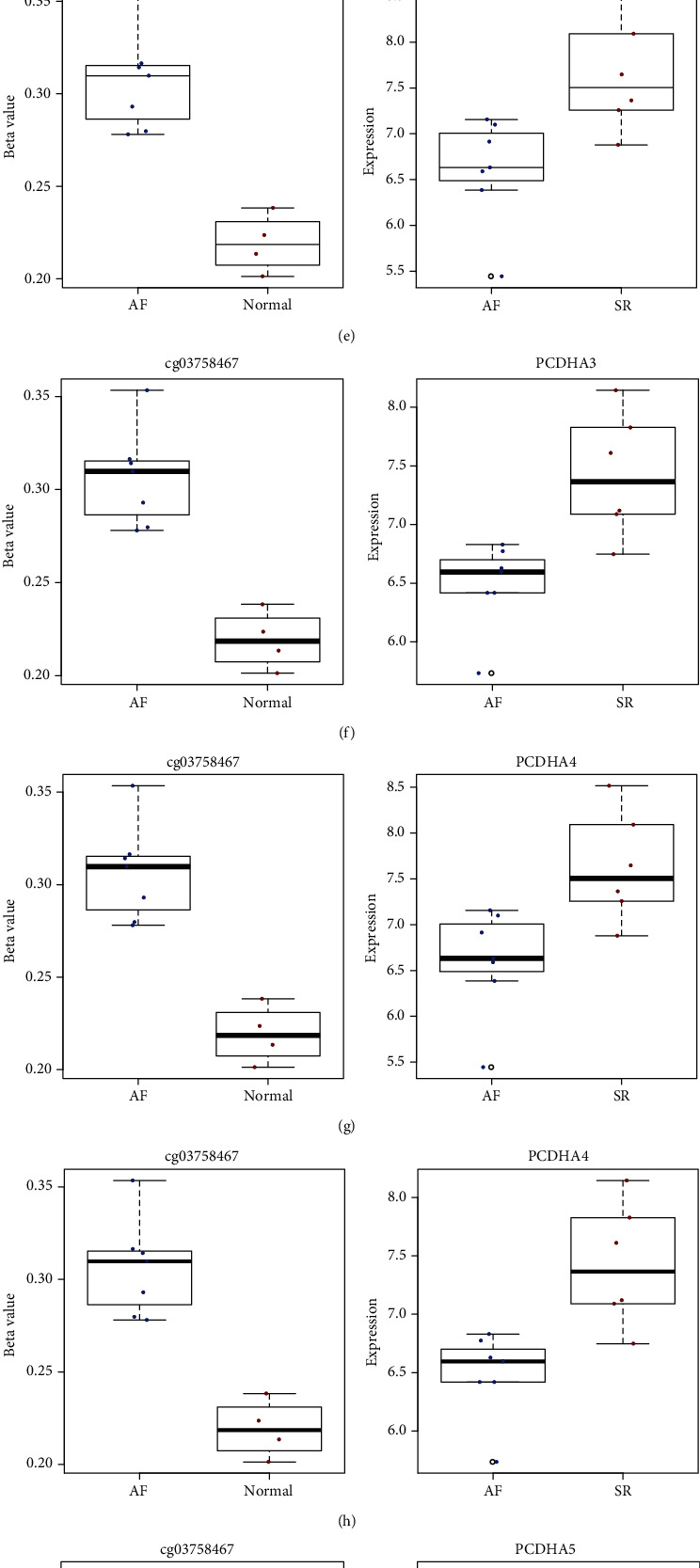
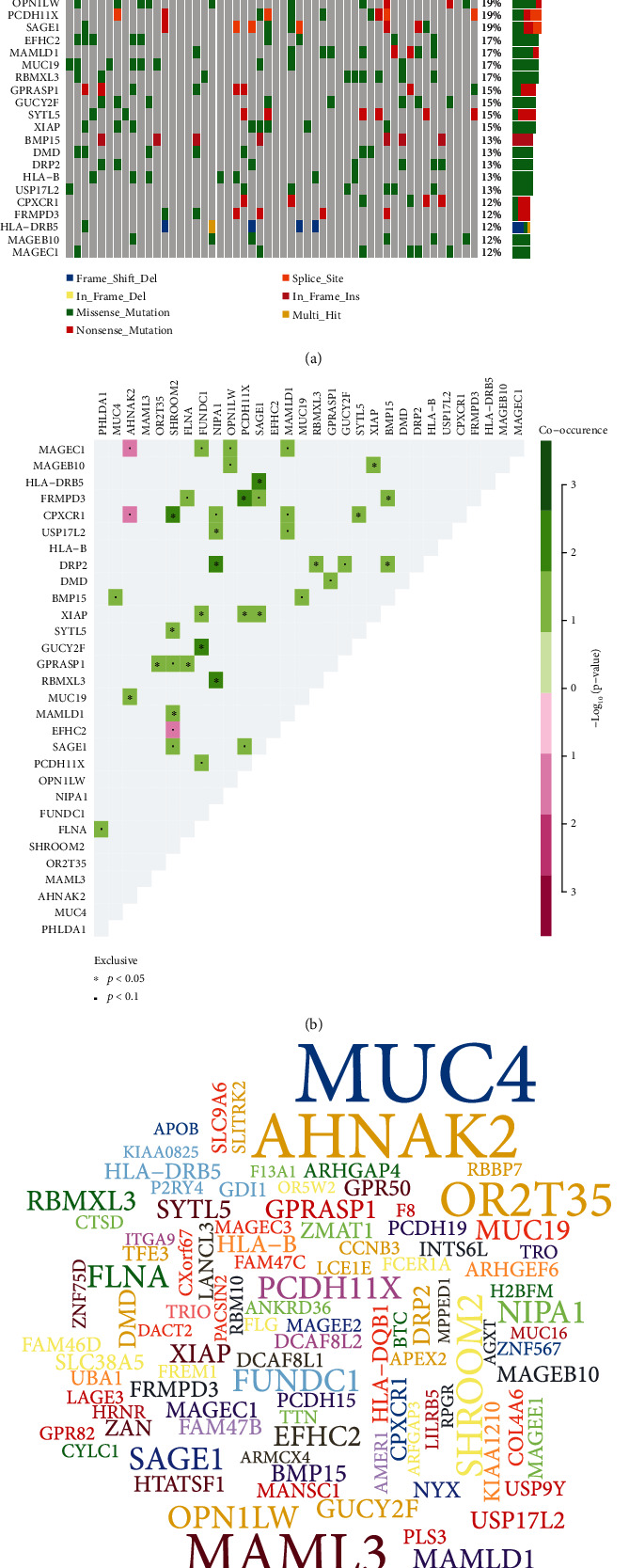
DEGs were identified between AF and controls, which were enriched in neutrophil activation and regulation of actin cytoskeleton. RHOA, CCR2, CASP8, and SYNPO2L exhibited abnormal expression and methylation, which have been confirmed to be related to AF. PCDHA family genes had high methylation and low expression in AF. We constructed two AF-related coexpression modules. Single-nucleotide polymorphism (SNP) was the most common mutation type in AF, especially T > C. MUC4 was the most frequent mutation gene, followed by PHLDA1, AHNAK2, and MAML3. There was no statistical difference in expression of AHNAK2 and MAML3, for AF. PHLDA1 and MUC4 were confirmed to be abnormally expressed in AF.
Our findings identified DEGs related to DNA methylation and mutation for AF, which may offer possible therapeutic targets and a new insight into the pathogenesis of AF from a multiomics perspective.
本研究旨在通过多组学分析,了解心房颤动(AF)的分子机制,并探索潜在的治疗靶点。
从基因表达综合数据库(GEO)中检索 AF 患者的转录组学和甲基化数据。筛选 AF 与正常样本之间差异表达基因(DEGs)和差异甲基化位点。然后,鉴定 AF 中高表达且低甲基化和低表达且高甲基化基因。进行加权基因共表达网络分析(WGCNA)以构建 AF 相关共表达网络。使用 52 例 AF 血液样本进行全外显子测序。R 中的 maftools 包可视化突变。在 AF 中使用独立数据集验证关键基因。
在 AF 与对照组之间鉴定出 DEGs,这些基因富集于中性粒细胞激活和肌动蛋白细胞骨架调节。RHOA、CCR2、CASP8 和 SYNPO2L 表现出异常表达和甲基化,这些已被证实与 AF 相关。AF 中 PCDHA 家族基因呈现高甲基化和低表达。我们构建了两个 AF 相关的共表达模块。单核苷酸多态性(SNP)是 AF 中最常见的突变类型,尤其是 T > C。MUC4 是最常见的突变基因,其次是 PHLDA1、AHNAK2 和 MAML3。AF 中 AHNAK2 和 MAML3 的表达没有统计学差异。PHLDA1 和 MUC4 被证实异常表达于 AF。
我们的研究结果确定了与 AF 相关的 DNA 甲基化和突变的 DEGs,这可能为 AF 提供潜在的治疗靶点,并从多组学角度为 AF 的发病机制提供新的见解。