Computer Science and Engineering, Michigan State University, East Lansing, 48824, USA.
Electrical Engineering, City University of Hong Kong, Hong Kong, People's Republic of China.
BMC Genomics. 2021 Apr 9;22(1):251. doi: 10.1186/s12864-021-07468-7.
With the development of third-generation sequencing (TGS) technologies, people are able to obtain DNA sequences with lengths from 10s to 100s of kb. These long reads allow protein domain annotation without assembly, thus can produce important insights into the biological functions of the underlying data. However, the high error rate in TGS data raises a new challenge to established domain analysis pipelines. The state-of-the-art methods are not optimized for noisy reads and have shown unsatisfactory accuracy of domain classification in TGS data. New computational methods are still needed to improve the performance of domain prediction in long noisy reads.
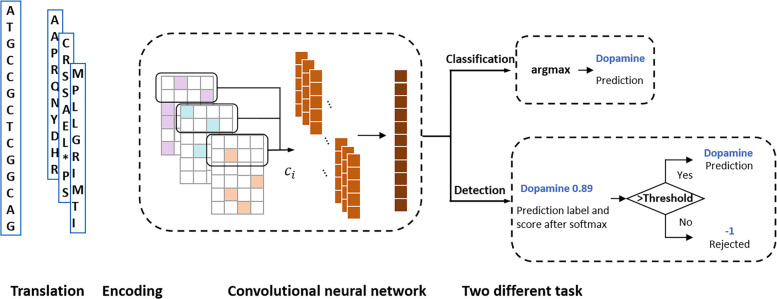
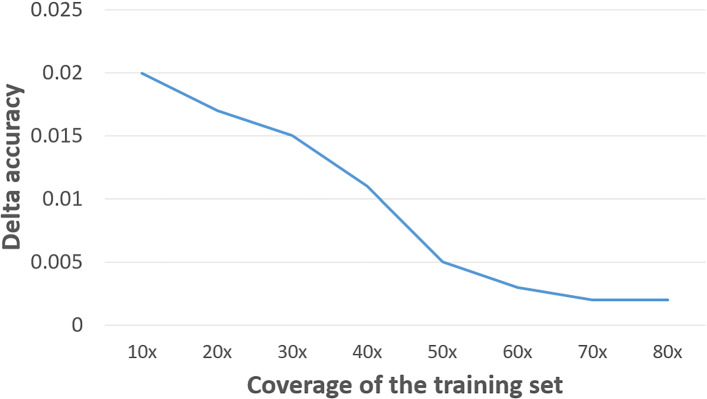
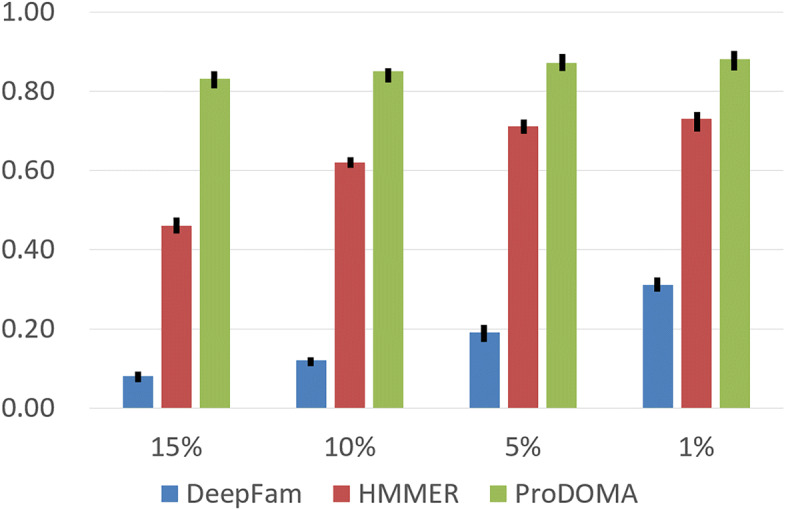
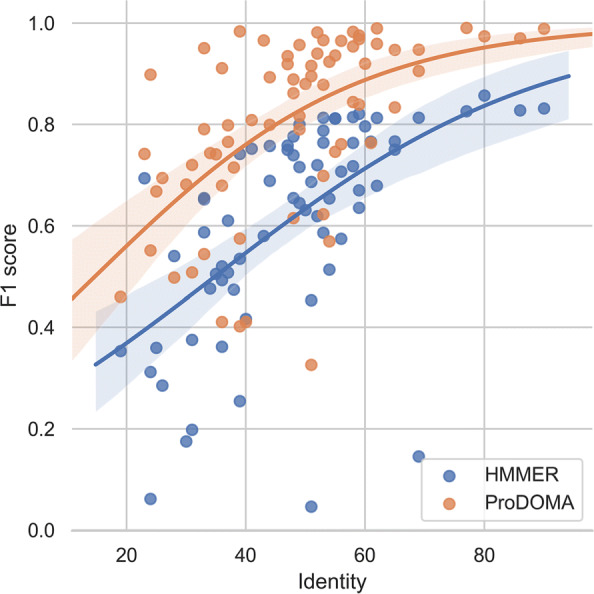
In this work, we introduce ProDOMA, a deep learning model that conducts domain classification for TGS reads. It uses deep neural networks with 3-frame translation encoding to learn conserved features from partially correct translations. In addition, we formulate our problem as an open-set problem and thus our model can reject reads not containing the targeted domains. In the experiments on simulated long reads of protein coding sequences and real TGS reads from the human genome, our model outperforms HMMER and DeepFam on protein domain classification.
In summary, ProDOMA is a useful end-to-end protein domain analysis tool for long noisy reads without relying on error correction.
随着第三代测序(TGS)技术的发展,人们能够获得长度从数十到数百 kb 的 DNA 序列。这些长读长允许在不进行组装的情况下注释蛋白质结构域,从而可以深入了解基础数据的生物学功能。然而,TGS 数据中的高错误率给已有的结构域分析流水线带来了新的挑战。最先进的方法并不是针对噪声读取进行优化的,并且在 TGS 数据中的结构域分类准确性上表现不佳。仍然需要新的计算方法来提高长噪声读取中的结构域预测性能。
在这项工作中,我们引入了 ProDOMA,这是一种针对 TGS 读取进行结构域分类的深度学习模型。它使用具有 3 帧翻译编码的深度神经网络,从部分正确的翻译中学习保守特征。此外,我们将我们的问题表述为开放集问题,因此我们的模型可以拒绝不包含目标结构域的读取。在对蛋白质编码序列的模拟长读取和人类基因组中的真实 TGS 读取的实验中,我们的模型在蛋白质结构域分类方面优于 HMMER 和 DeepFam。
总之,ProDOMA 是一种无需依赖纠错的有用的端到端长噪声读取蛋白质结构域分析工具。