Electrical Engineering, City University of Hong Kong, Hong Kong, China.
BMC Biol. 2021 Nov 24;19(1):250. doi: 10.1186/s12915-021-01180-4.
Prokaryotic viruses, which infect bacteria and archaea, are the most abundant and diverse biological entities in the biosphere. To understand their regulatory roles in various ecosystems and to harness the potential of bacteriophages for use in therapy, more knowledge of viral-host relationships is required. High-throughput sequencing and its application to the microbiome have offered new opportunities for computational approaches for predicting which hosts particular viruses can infect. However, there are two main challenges for computational host prediction. First, the empirically known virus-host relationships are very limited. Second, although sequence similarity between viruses and their prokaryote hosts have been used as a major feature for host prediction, the alignment is either missing or ambiguous in many cases. Thus, there is still a need to improve the accuracy of host prediction.
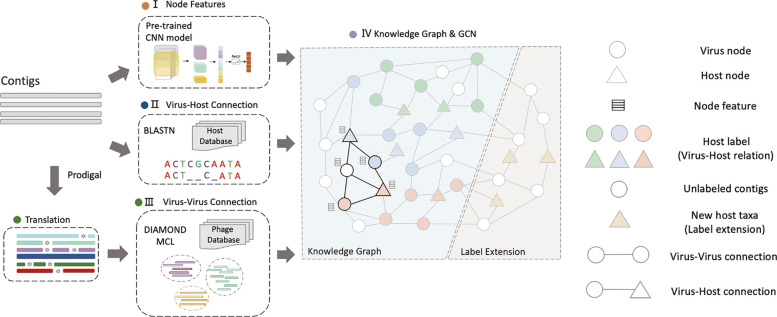
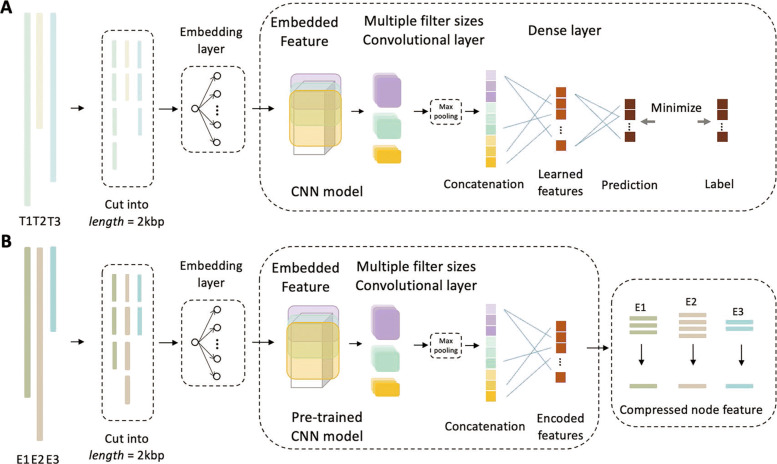
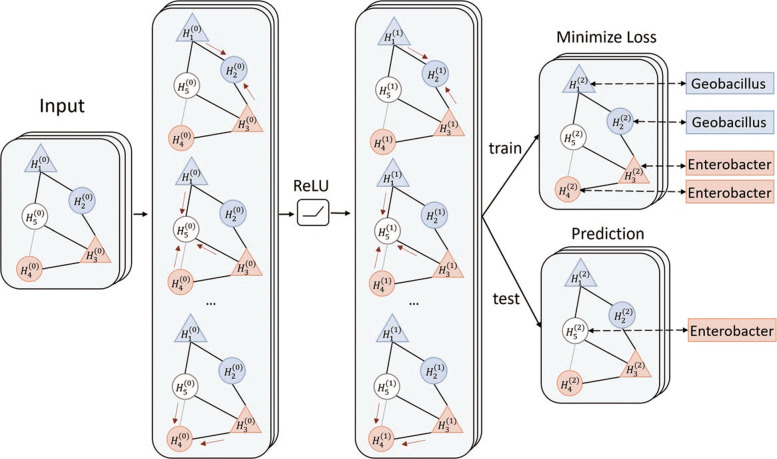
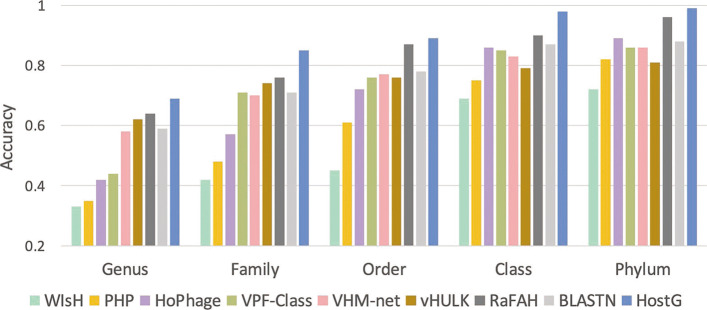
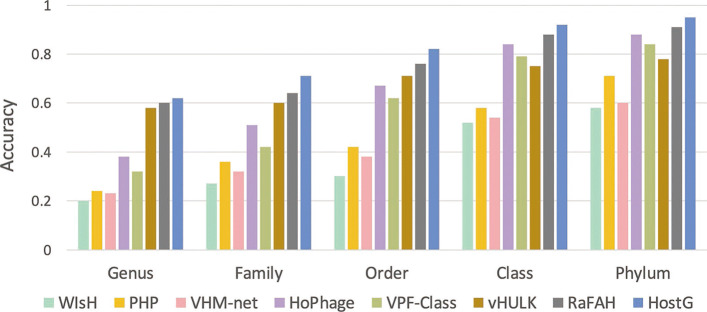
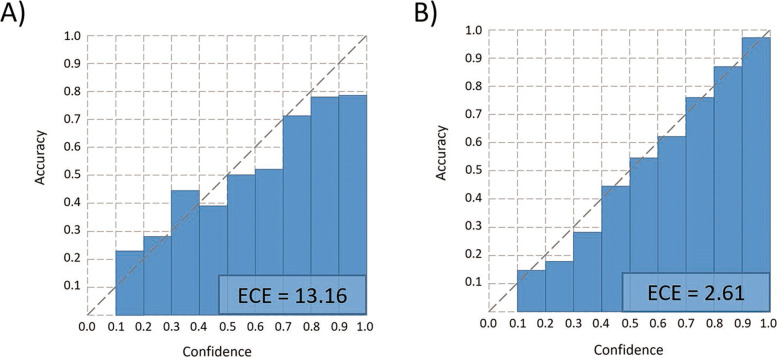
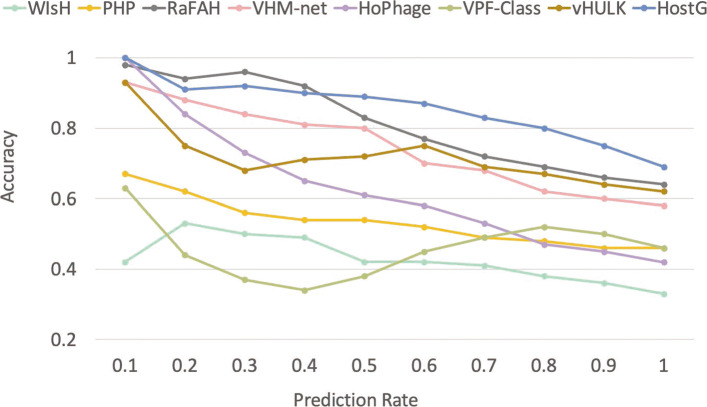
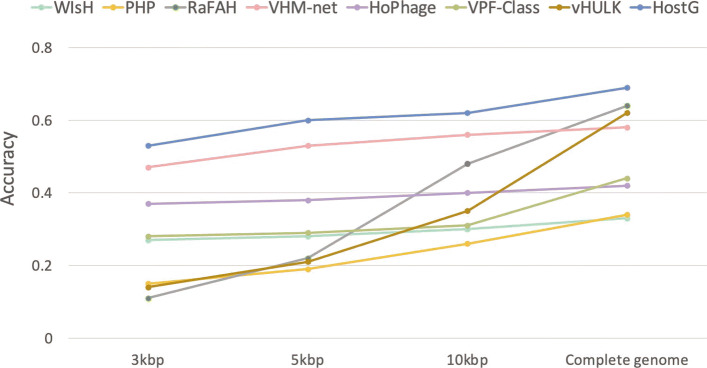
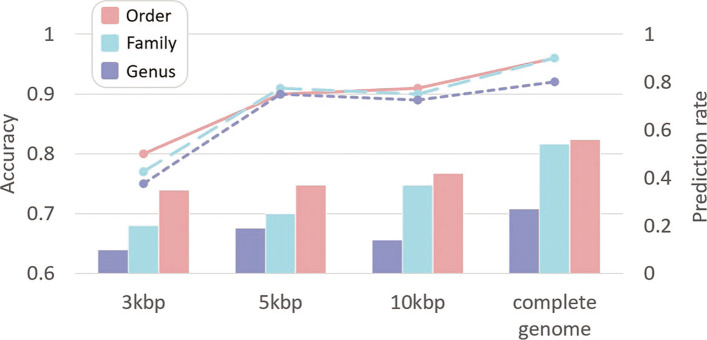
In this work, we present a semi-supervised learning model, named HostG, to conduct host prediction for novel viruses. We construct a knowledge graph by utilizing both virus-virus protein similarity and virus-host DNA sequence similarity. Then graph convolutional network (GCN) is adopted to exploit viruses with or without known hosts in training to enhance the learning ability. During the GCN training, we minimize the expected calibrated error (ECE) to ensure the confidence of the predictions. We tested HostG on both simulated and real sequencing data and compared its performance with other state-of-the-art methods specifically designed for virus host classification (VHM-net, WIsH, PHP, HoPhage, RaFAH, vHULK, and VPF-Class).
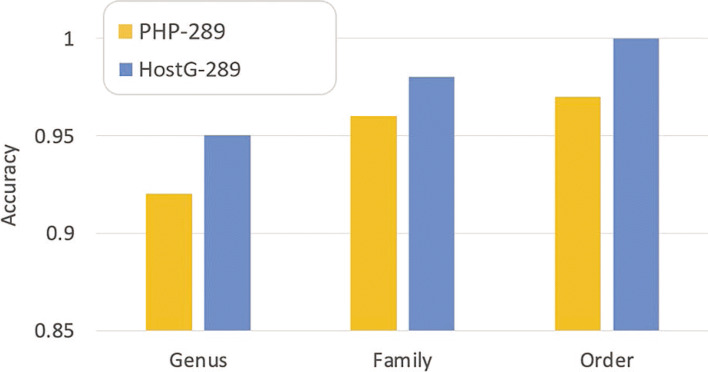
HostG outperforms other popular methods, demonstrating the efficacy of using a GCN-based semi-supervised learning approach. A particular advantage of HostG is its ability to predict hosts from new taxa.
感染细菌和古菌的原核病毒是生物圈中最丰富和最多样化的生物实体。为了了解它们在各种生态系统中的调节作用,并利用噬菌体的潜力进行治疗,我们需要更多地了解病毒-宿主关系。高通量测序及其在微生物组中的应用为预测特定病毒可以感染哪些宿主的计算方法提供了新的机会。然而,计算宿主预测存在两个主要挑战。首先,经验上已知的病毒-宿主关系非常有限。其次,尽管病毒与其原核宿主之间的序列相似性已被用作宿主预测的主要特征,但在许多情况下,对齐要么缺失要么模糊。因此,仍然需要提高宿主预测的准确性。
在这项工作中,我们提出了一种半监督学习模型,称为 HostG,用于对新病毒进行宿主预测。我们通过利用病毒-病毒蛋白相似性和病毒-宿主 DNA 序列相似性构建知识图谱。然后采用图卷积网络(GCN)来利用具有或不具有已知宿主的病毒进行训练,以增强学习能力。在 GCN 训练过程中,我们最小化期望校准误差(ECE)以确保预测的置信度。我们在模拟和真实测序数据上测试了 HostG,并将其性能与专门为病毒宿主分类设计的其他最先进的方法(VHM-net、WIsH、PHP、HoPhage、RaFAH、vHULK 和 VPF-Class)进行了比较。
HostG 优于其他流行方法,证明了基于 GCN 的半监督学习方法的有效性。HostG 的一个特别优势是它能够从新的分类单元预测宿主。