Bilalbegović Goranka, Maksimović Aleksandar, Valencic Lynne A, Lehtola Susi
Department of Physics, Faculty of Science, University of Zagreb, Bijenička cesta 32, 10000 Zagreb, Croatia.
Center of Excellence for Advanced Materials and Sensing Devices, Rudjer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia.
ACS Earth Space Chem. 2021 Mar 18;5(3):436-448. doi: 10.1021/acsearthspacechem.0c00238. Epub 2021 Feb 24.
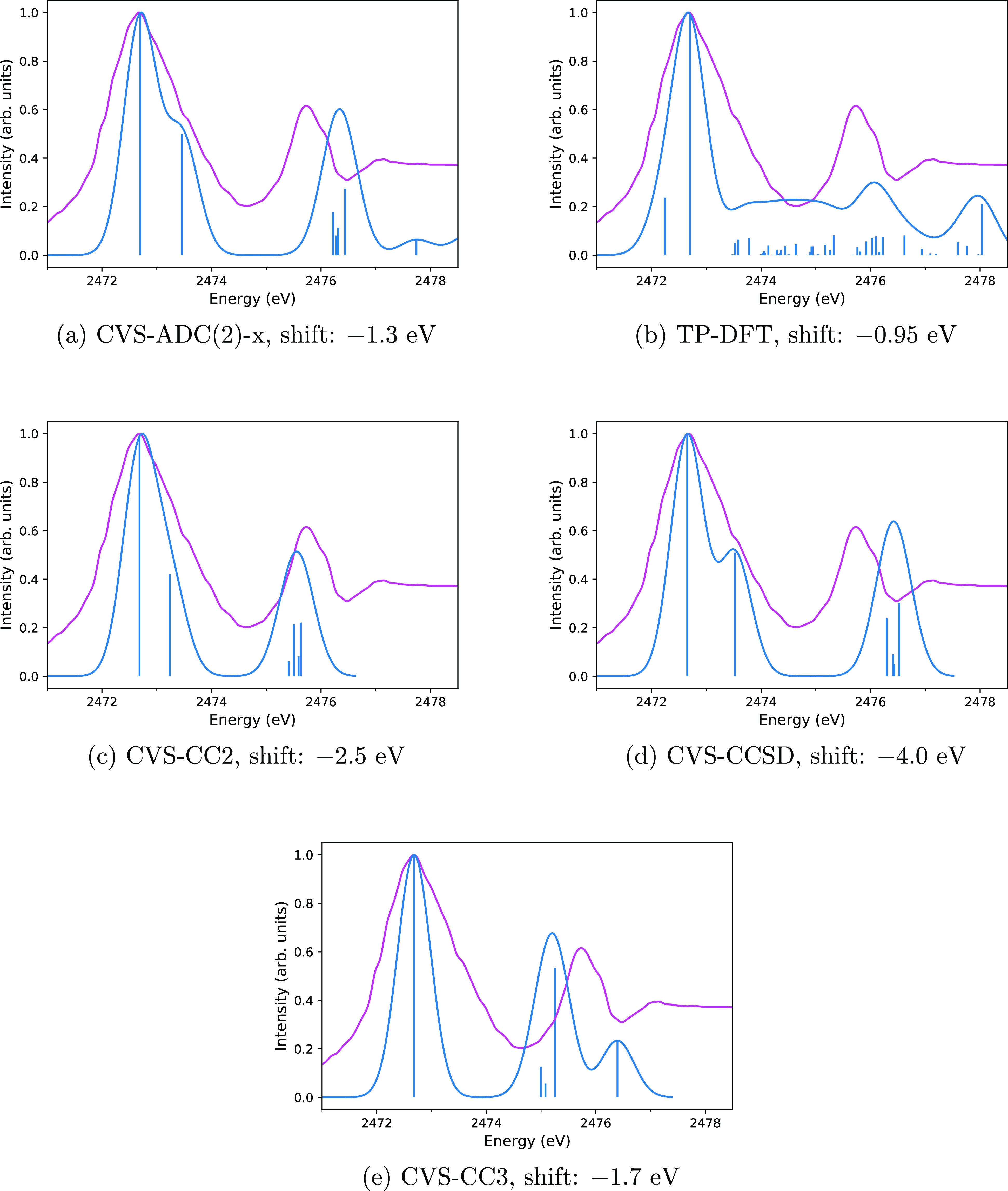
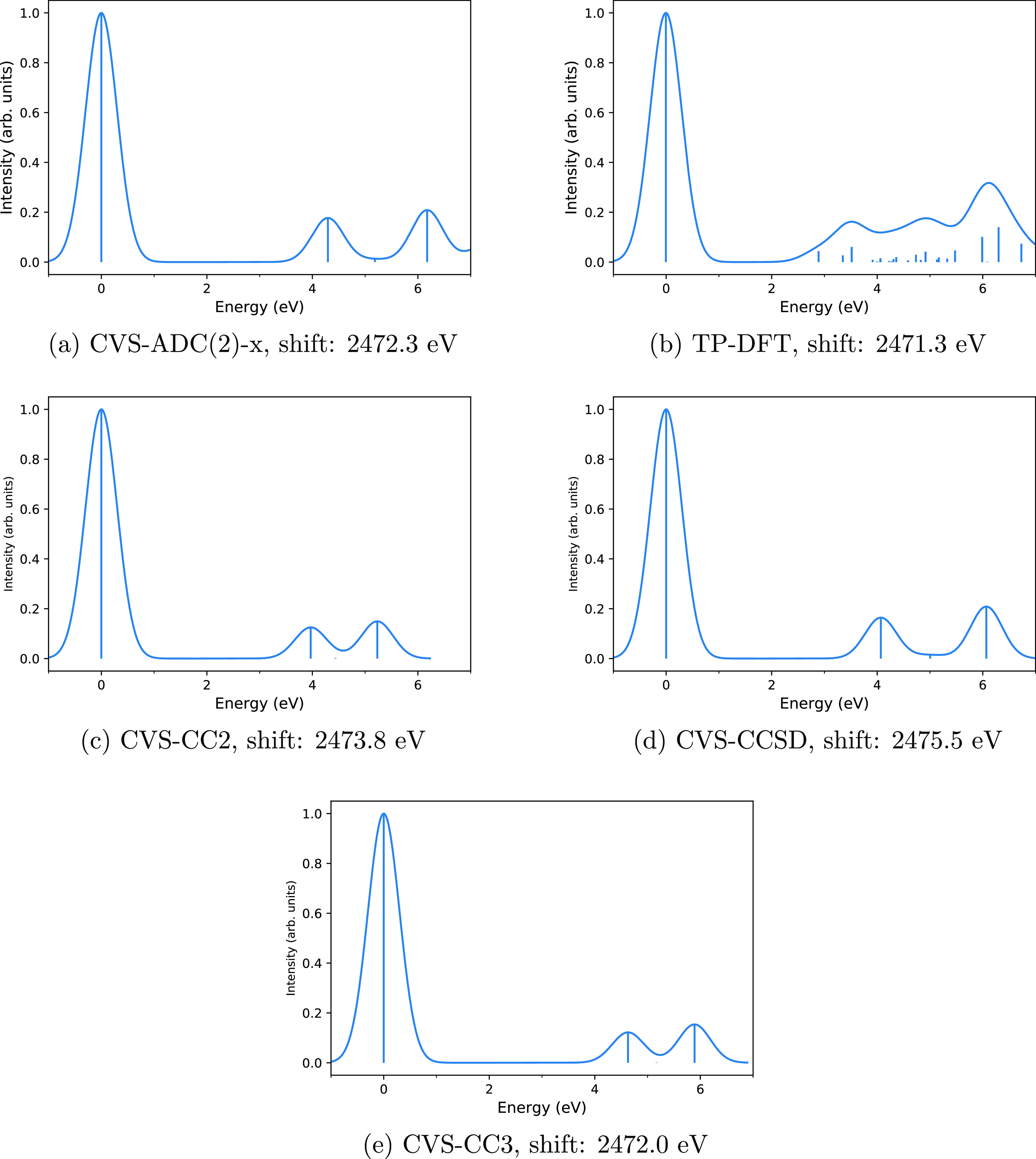
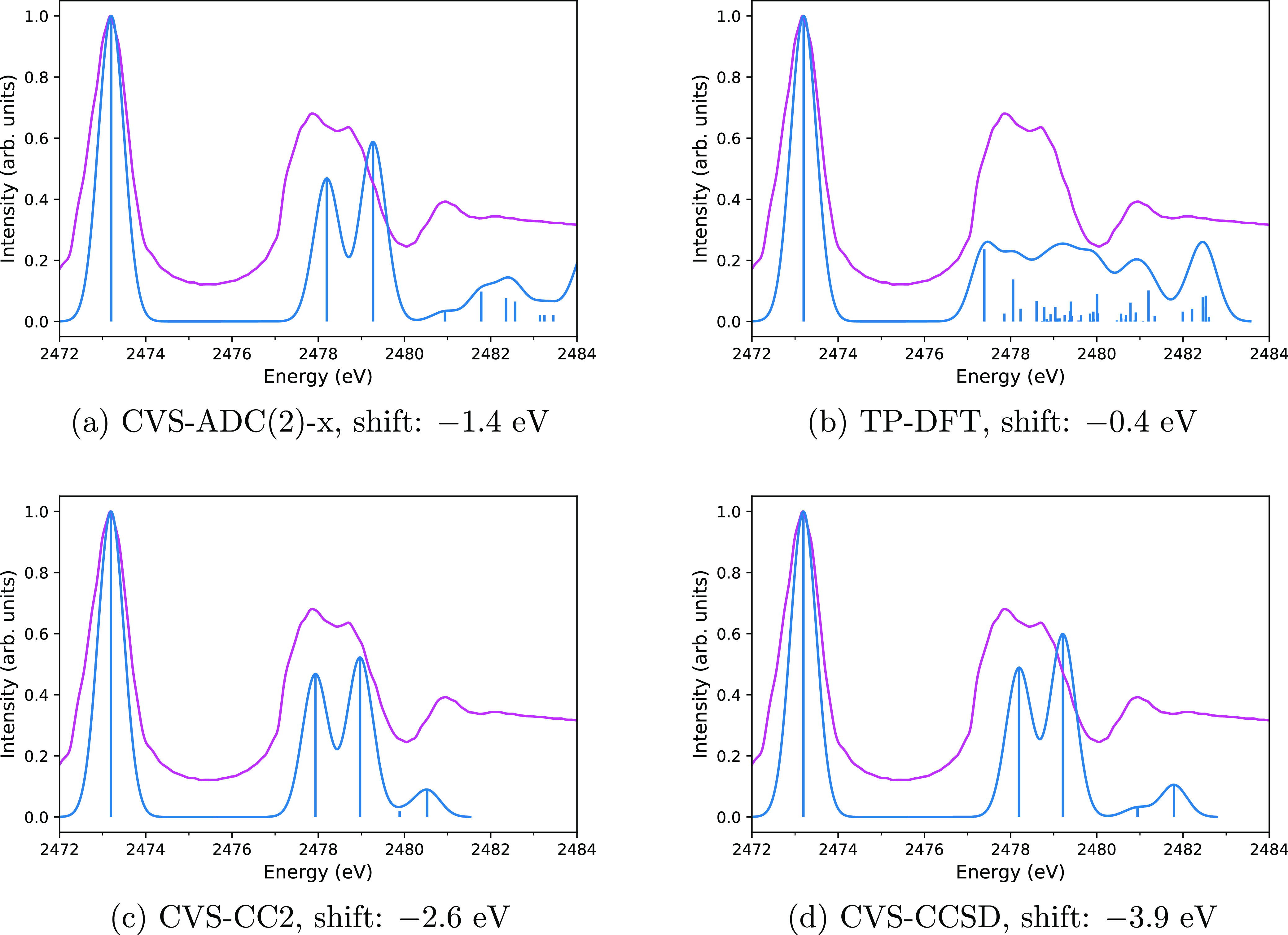
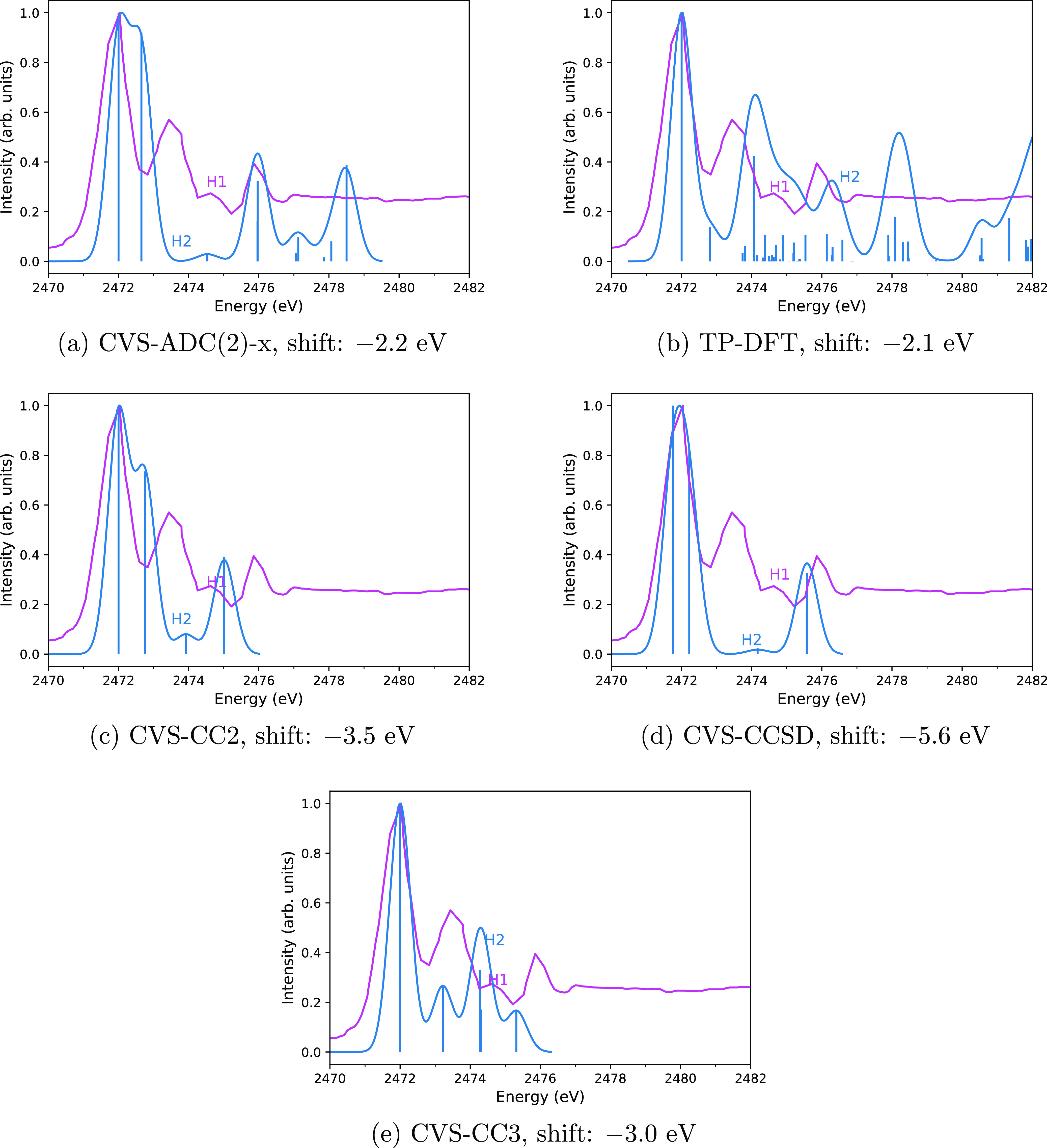
X-ray astronomy lacks high resolution spectra of interstellar dust analogues and molecules, severely hampering interstellar medium studies based on upcoming X-ray missions. Various theoretical approaches may be used to address this problem, but they must first be shown to reproduce reliable spectra compared to the experiment. In this work, we calculate the sulfur K edge X-ray absorption spectra of HS, SO, and OCS, whose spectra are already known from X-ray experiments and predict the X-ray spectrum of CS, which as far as we are aware has not been measured, thereby hampering its detection by X-ray telescopes. We chose these four molecules as the astrochemistry of sulfur is an unsolved problem and as the four molecules are already known to exist in space. We consider three types of methods for modeling the X-ray spectra: more accurate calculations with the algebraic-diagrammatic construction (ADC) and the CC2, CCSD, and CC3 coupled cluster (CC) approaches as well as more affordable ones with transition potential density functional theory (TP-DFT). A comparison of our computational results to previously reported experimental spectra shows that the core-valence separation (CVS) approaches CVS-ADC(2)-x and CVS-CC3 generally yield a good qualitative level of agreement with the experiment, suggesting that they can be used for interpreting measured spectra, while the TP-DFT method is not reliable for these molecules. However, quantitative agreement with the experiment is still outside the reach of the computational methods studied in this work.
X射线天文学缺乏星际尘埃类似物和分子的高分辨率光谱,这严重阻碍了基于即将开展的X射线任务的星际介质研究。可以采用各种理论方法来解决这个问题,但首先必须证明它们与实验相比能够重现可靠的光谱。在这项工作中,我们计算了HS、SO和OCS的硫K边X射线吸收光谱,这些光谱已经通过X射线实验得知,并且预测了CS的X射线光谱,据我们所知,CS的X射线光谱尚未被测量,因此阻碍了通过X射线望远镜对其进行探测。我们选择这四种分子是因为硫的天体化学是一个尚未解决的问题,并且这四种分子在太空中已经被证实存在。我们考虑了三种对X射线光谱进行建模的方法:使用代数图构建(ADC)以及CC2、CCSD和CC3耦合簇(CC)方法进行更精确的计算,以及使用过渡势密度泛函理论(TP-DFT)进行成本更低的计算。将我们的计算结果与先前报道的实验光谱进行比较表明,芯价分离(CVS)方法CVS-ADC(2)-x和CVS-CC3通常与实验在定性水平上有较好的一致性,这表明它们可用于解释测量光谱,而TP-DFT方法对于这些分子不可靠。然而,与实验的定量一致性仍然超出了本工作所研究的计算方法的能力范围。