Hosseini Maryam, Chen Wanqiu, Xiao Daliao, Wang Charles
Center for Genomics, School of Medicine, Loma Linda University, Loma Linda, CA 92350, USA.
Lawrence D. Longo, MD Center for Perinatal Biology, Department of Basic Sciences, School of Medicine, Loma Linda University, Loma Linda, CA 92350, USA.
Precis Clin Med. 2021 Jan 18;4(1):1-16. doi: 10.1093/pcmedi/pbab001. eCollection 2021 Mar.
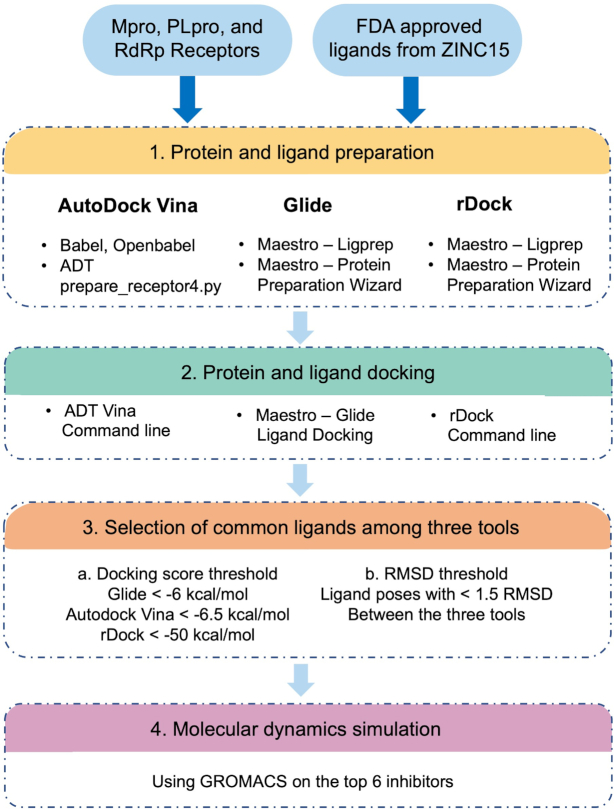
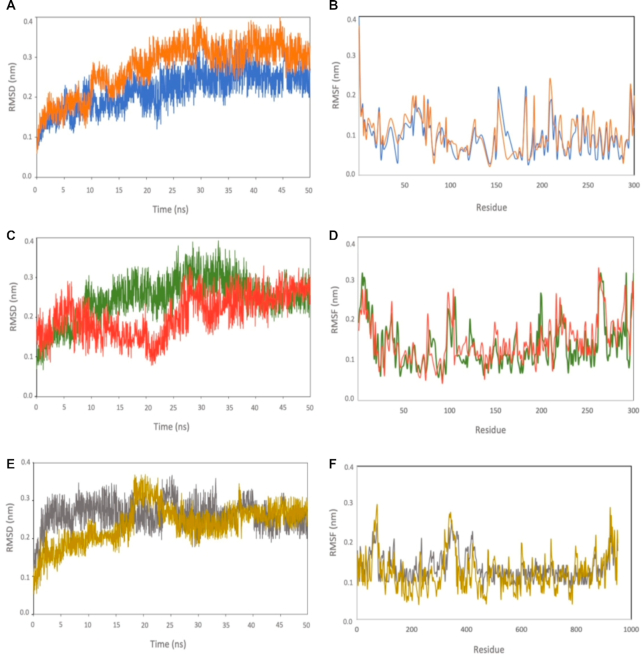
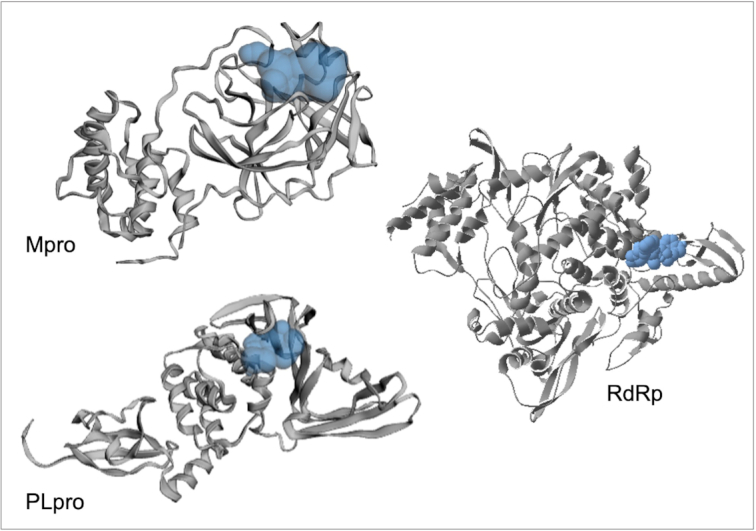
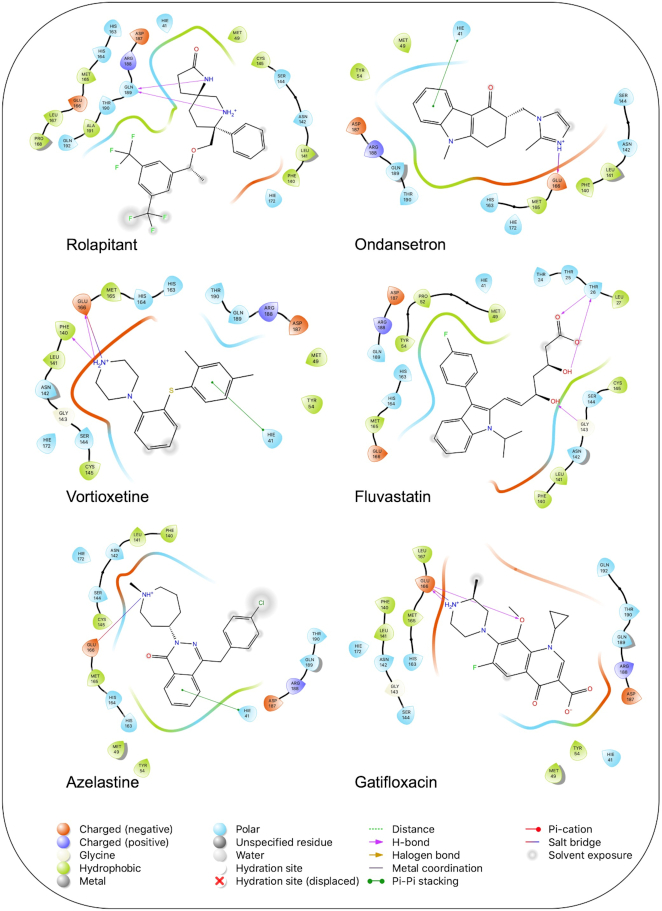
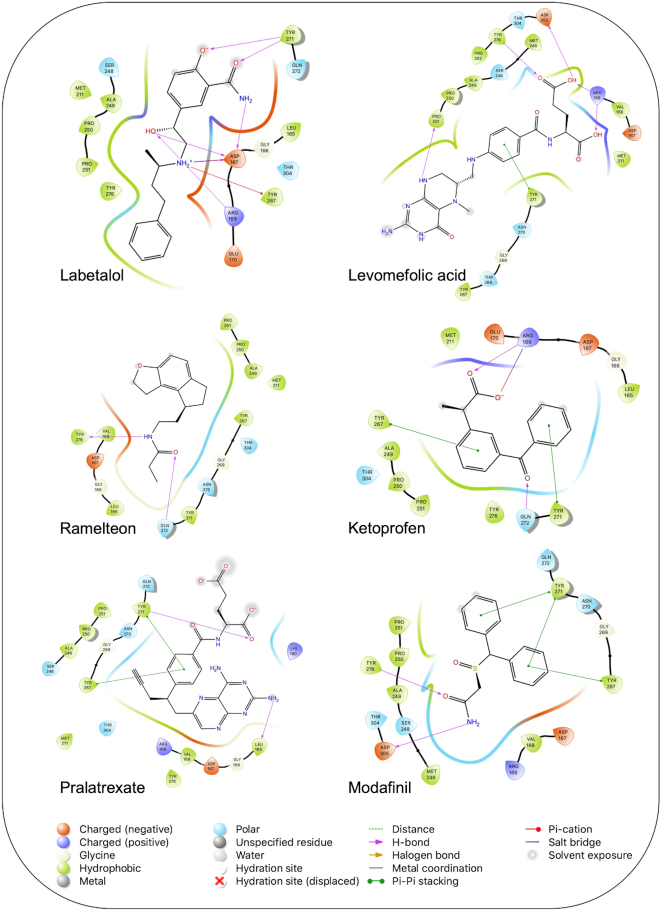
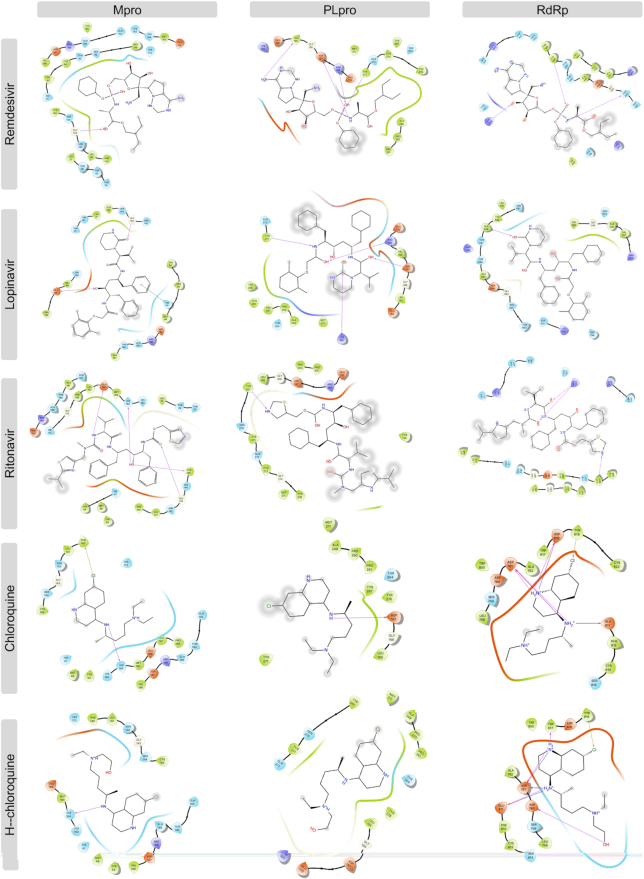
The pandemic of novel coronavirus disease 2019 (COVID-19) has rampaged the world, with more than 58.4 million confirmed cases and over 1.38 million deaths across the world by 23 November 2020. There is an urgent need to identify effective drugs and vaccines to fight against the virus. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the family of coronaviruses consisting of four structural and 16 non-structural proteins (NSP). Three non-structural proteins, main protease (Mpro), papain-like protease (PLpro), and RNA-dependent RNA polymerase (RdRp), are believed to have a crucial role in replication of the virus. We applied computational ligand-receptor binding modeling and performed comprehensive virtual screening on FDA-approved drugs against these three SARS-CoV-2 proteins using AutoDock Vina, Glide, and rDock. Our computational studies identified six novel ligands as potential inhibitors against SARS-CoV-2, including antiemetics rolapitant and ondansetron for Mpro; labetalol and levomefolic acid for PLpro; and leucal and antifungal natamycin for RdRp. Molecular dynamics simulation confirmed the stability of the ligand-protein complexes. The results of our analysis with some other suggested drugs indicated that chloroquine and hydroxychloroquine had high binding energy (low inhibitory effect) with all three proteins-Mpro, PLpro, and RdRp. In summary, our computational molecular docking approach and virtual screening identified some promising candidate SARS-CoV-2 inhibitors that may be considered for further clinical studies.
2019年新型冠状病毒病(COVID-19)大流行肆虐全球,截至2020年11月23日,全球确诊病例超过5840万例,死亡人数超过138万。迫切需要确定有效的药物和疫苗来对抗该病毒。严重急性呼吸综合征冠状病毒2(SARS-CoV-2)属于冠状病毒科,由四种结构蛋白和16种非结构蛋白(NSP)组成。三种非结构蛋白,即主要蛋白酶(Mpro)、木瓜样蛋白酶(PLpro)和RNA依赖性RNA聚合酶(RdRp),被认为在病毒复制中起关键作用。我们应用计算配体-受体结合模型,使用AutoDock Vina、Glide和rDock对FDA批准的针对这三种SARS-CoV-2蛋白的药物进行了全面的虚拟筛选。我们的计算研究确定了六种新型配体作为SARS-CoV-2的潜在抑制剂,包括用于Mpro的止吐药罗拉匹坦和昂丹司琼;用于PLpro的拉贝洛尔和左亚叶酸;以及用于RdRp的亮氨酸和抗真菌药游霉素。分子动力学模拟证实了配体-蛋白质复合物的稳定性。我们对其他一些建议药物的分析结果表明,氯喹和羟氯喹与所有三种蛋白——Mpro、PLpro和RdRp——具有高结合能(低抑制作用)。总之,我们的计算分子对接方法和虚拟筛选确定了一些有前景的SARS-CoV-2抑制剂候选物,可考虑用于进一步的临床研究。