Department of Neuroscience, Università Cattolica del Sacro Cuore, Facoltà di medicina e Chirurgia, Rome, Italy.
UOC Neurologia, Policlinico Universitario A.Gemelli IRCCS, Rome, Italy.
Eur J Neurol. 2021 Aug;28(8):2784-2788. doi: 10.1111/ene.14868. Epub 2021 May 27.
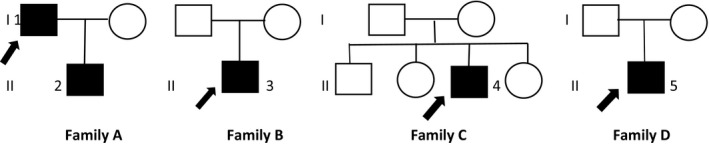
Spinocerebellar ataxia 21 (SCA21) is a rare autosomal dominant neurodegenerative disorder caused by TMEM240 gene mutations. To date, SCA21 has been reported only in a limited number of families worldwide. Here, we describe clinical and molecular findings in five additional SCA21 patients from four unrelated families, diagnosed through a multicentre next generation sequencing-based molecular screening project on a large cohort of patients with degenerative and congenital ataxias.
A cohort of 393 patients with ataxia of unknown aetiology was selected. Following the identification of heterozygous pathogenic TMEM240 variants using a target resequencing panel, we carried out an in-depth phenotyping of the novel SCA21 patients.
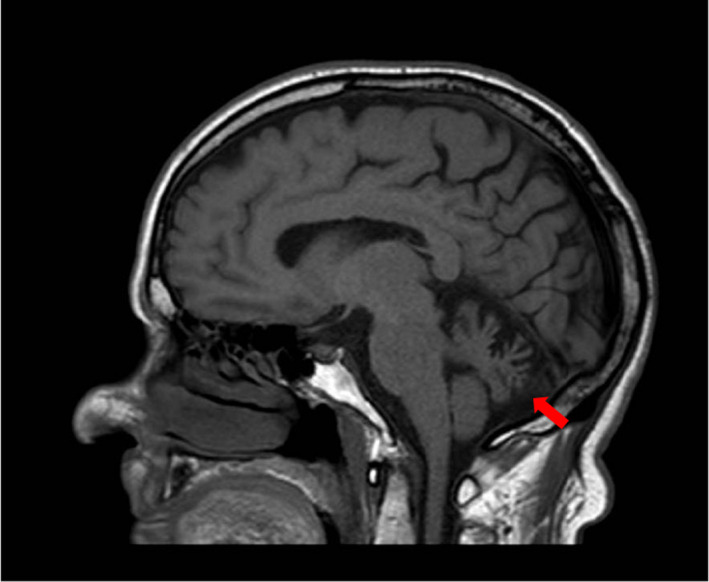
Five patients from four unrelated families, three of Italian and one of Libyan origin, were identified. These patients were carriers of previously reported TMEM240 mutations. Clinically, our SCA21 cohort includes both adult onset, slowly progressive cerebellar ataxias associated with cognitive impairment resembling cerebellar cognitive affective syndrome and early onset forms associated with cognitive delay, neuropsychiatric features, or evidence of hypomyelination on brain magnetic resonance imaging. None of our patients exhibited signs of extrapyramidal involvement. The so-called "recurrent" c.509C>T (p.Pro170Leu) mutation was detected in two of four families, corroborating its role as a hot spot.
Our results confirm that SCA21 is present also in Italy, suggesting that it might not be as rare as previously thought. The phenotype of these novel SCA21 patients indicates that slowly progressive cerebellar ataxia, and cognitive and psychiatric symptoms are the most typical clinical features associated with mutations in the TMEM240 gene.
脊髓小脑性共济失调 21 型(SCA21)是一种罕见的常染色体显性遗传性神经退行性疾病,由 TMEM240 基因突变引起。迄今为止,SCA21 仅在全球少数几个家族中被报道过。在这里,我们描述了另外五个来自四个不相关家族的 SCA21 患者的临床和分子发现,这些患者是通过对一组患有退行性和先天性共济失调的患者进行基于下一代测序的分子筛查项目在一个大的患者队列中诊断出来的。
我们选择了一组 393 名病因不明的共济失调患者。在使用靶向重测序面板鉴定出杂合致病性 TMEM240 变体后,我们对新的 SCA21 患者进行了深入的表型分析。
从四个不相关的家族中发现了五名患者,其中三名来自意大利,一名来自利比亚。这些患者携带了先前报道的 TMEM240 突变。临床上,我们的 SCA21 患者队列包括与小脑认知情感综合征相似的认知障碍相关的成人起病、缓慢进展的小脑共济失调,以及与认知延迟、神经精神特征或磁共振成像上的脱髓鞘证据相关的早发型形式。我们的患者均无锥体外系受累的迹象。在四个家族中的两个家族中检测到了所谓的“复发性”c.509C>T(p.Pro170Leu)突变,证实了其作为热点的作用。
我们的结果证实 SCA21 也存在于意大利,表明其可能不像以前认为的那样罕见。这些新的 SCA21 患者的表型表明,缓慢进展的小脑共济失调、认知和精神症状是与 TMEM240 基因突变相关的最典型的临床特征。