Thorstensen Matt J, Baerwald Melinda R, Jeffries Ken M
Department of Biological Sciences, University of Manitoba, Winnipeg, MB, R3T 2N2, Canada.
California Department of Water Resources, West Sacramento, CA, 95691, USA.
BMC Genomics. 2021 Apr 15;22(1):273. doi: 10.1186/s12864-021-07592-4.
Messenger RNA sequencing is becoming more common in studies of non-model species and is most often used for gene expression-based investigations. However, the method holds potential for numerous other applications as well-including analyses of alternative splicing, population structure, and signatures of selection. To maximize the utility of mRNA data sets, distinct analyses may be combined such as by exploring dynamics between gene expression with signatures of selection in the context of population structure. Here, we compare two published data sets describing two populations of a minnow species endemic to the San Francisco Estuary (Sacramento splittail, Pogonichthys macrolepidotus): a microsatellite data set showing population structure, and an mRNA whole transcriptome data set obtained after the two populations were exposed to a salinity challenge. We compared measures of population structure and genetic variation using single nucleotide polymorphisms (SNPs) called from mRNA from the whole transcriptome sequencing study with those patterns determined from microsatellites. For investigating plasticity and evolution, intra- and inter-population transcriptome plasticity was investigated with differential gene expression, differential exon usage, and gene expression variation. Outlier SNP analysis was also performed on the mRNA data set and signatures of selection and phenotypic plasticity were investigated on an individual-gene basis.
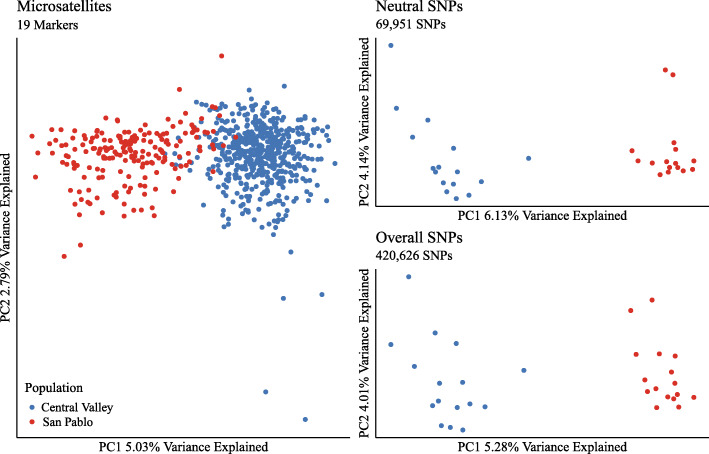
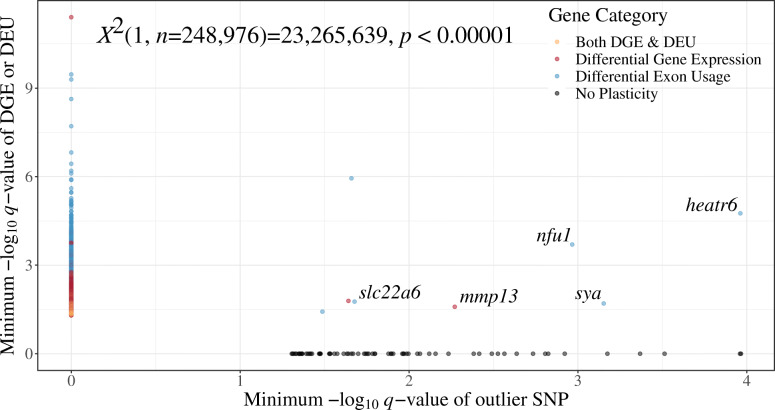
We found that mRNA sequencing revealed patterns of population structure consistent with those found with microsatellites, but with lower magnitudes of genetic variation and population differentiation consistent with widespread purifying selection expected when using mRNA. In addition, within individual genes, phenotypic plasticity or signatures of selection were found in almost mutual exclusion (except heatr6, nfu1, slc22a6, sya, and mmp13).
These results show that an mRNA sequencing data set may have multiple uses, including describing population structure and for investigating the mechanistic interplay of evolution and plasticity in adaptation. MRNA sequencing thus complements traditional sequencing methods used for population genetics, in addition to its utility for describing phenotypic plasticity.
信使核糖核酸测序在非模式物种研究中越来越普遍,最常用于基于基因表达的研究。然而,该方法也有许多其他潜在应用,包括可变剪接分析、群体结构分析和选择特征分析。为了最大限度地利用信使核糖核酸数据集,可以将不同的分析方法结合起来,例如在群体结构背景下,通过探索基因表达与选择特征之间的动态关系。在这里,我们比较了两个已发表的数据集,这两个数据集描述了旧金山河口特有的一种米诺鱼(萨克拉门托裂尾鱼,Pogonichthys macrolepidotus)的两个种群:一个显示群体结构的微卫星数据集,以及两个种群在盐度挑战后获得的信使核糖核酸全转录组数据集。我们使用全转录组测序研究中从信使核糖核酸中鉴定出的单核苷酸多态性(SNP),将群体结构和遗传变异的测量结果与从微卫星中确定的模式进行了比较。为了研究可塑性和进化,我们通过差异基因表达、差异外显子使用和基因表达变异,研究了种群内和种群间的转录组可塑性。我们还对信使核糖核酸数据集进行了异常SNP分析,并在个体基因水平上研究了选择特征和表型可塑性。
我们发现,信使核糖核酸测序揭示的群体结构模式与微卫星分析的结果一致,但遗传变异程度和群体分化程度较低,这与使用信使核糖核酸时预期的广泛纯化选择一致。此外,在单个基因中,表型可塑性或选择特征几乎相互排斥(除了heatr6、nfu1、slc22a6、sya和mmp13)。
这些结果表明,信使核糖核酸测序数据集可能有多种用途,包括描述群体结构以及研究进化和可塑性在适应过程中的机制相互作用。因此,信使核糖核酸测序除了可用于描述表型可塑性外,还补充了用于群体遗传学的传统测序方法。