He Shun, Lu Yuanyuan, Guo Yuetong, Li Shijin, Lu Xiao, Shao Shuai, Zhou Handan, Wang Ruiqi, Wang Jiguang, Gao Pingjin, Li Xiaodong
Department of Cardiovascular Medicine, Ruijin Hospital and State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Shanghai Institute of Hypertension, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Department of Hypertension, Ruijin Hospital and State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Shanghai Institute of Hypertension, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Front Cell Dev Biol. 2021 Apr 1;9:644954. doi: 10.3389/fcell.2021.644954. eCollection 2021.
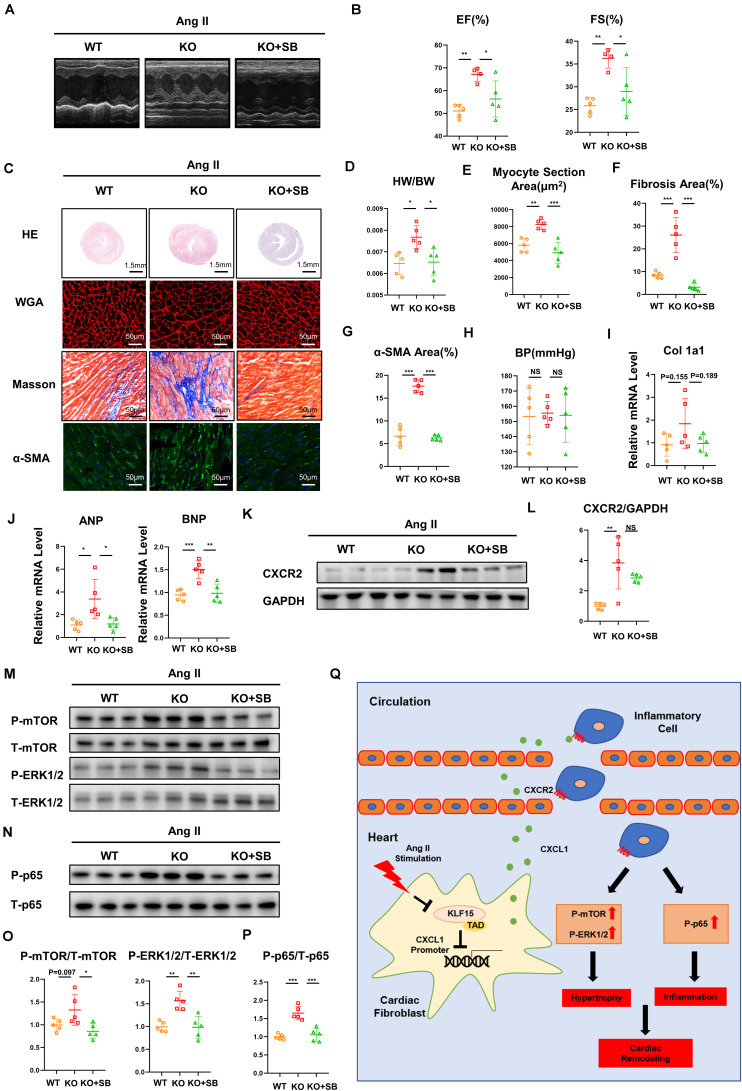
Inflammation is involved in cardiac remodeling. In response to pathological stimuli, activated cardiac fibroblasts (CFs) secreting inflammatory cytokines and chemokines play an important role in monocyte/macrophage recruitment. However, the precise mechanism of CF-mediated inflammatory response in hypertension-induced cardiac remodeling remains unclear. In the present study, we investigated the role of transcription factor Krüppel-like factor 15 (KLF15) in this process. We found that KLF15 expression decreased while chemokine and its receptor expression increased in the hearts of angiotensin II (Ang II)-infused mice. Compared to the wild-type mice, KLF15 knockout (KO) mice aggravated Ang II-induced cardiac hypertrophy and fibrosis. Deficiency of KLF15 promoted macrophage accumulation, increase of and expression, and mTOR, ERK1/2, NF-κB-p65 signaling activation in the hearts. Mechanistically, Ang II dose- dependently decreased KLF15 expression and increased secretion from cardiac fibroblasts but not cardiac myoblasts. Loss- or gain-of-function studies have shown that KLF15 negatively regulated expression through its transactivation domain (TAD). Intriguingly, the adenovirus-mediated full length of KLF15-but not KLF15 with TAD deletion overexpression-markedly prevented pathological change in Ang II-infused mice. Notably, the administration of inhibitor SB265610 reversed KLF15 knockout-mediated aggravation of cardiac dysfunction, remodeling, and inflammation induced by Ang II. In conclusion, our study identifies that KLF15 in cardiac fibroblasts negatively regulates axis-mediated inflammatory response and subsequent cardiac remodeling in hypertension.
炎症参与心脏重塑。响应病理刺激,分泌炎性细胞因子和趋化因子的活化心脏成纤维细胞(CFs)在单核细胞/巨噬细胞募集中起重要作用。然而,在高血压诱导的心脏重塑中,CF介导的炎症反应的确切机制仍不清楚。在本研究中,我们调查了转录因子Krüppel样因子15(KLF15)在此过程中的作用。我们发现,在输注血管紧张素II(Ang II)的小鼠心脏中,KLF15表达降低,而趋化因子及其受体表达增加。与野生型小鼠相比,KLF15基因敲除(KO)小鼠加重了Ang II诱导的心脏肥大和纤维化。KLF15的缺乏促进了巨噬细胞的积累、 和 表达的增加以及心脏中mTOR、ERK1/2、NF-κB-p65信号的激活。机制上,Ang II剂量依赖性地降低KLF15表达并增加心脏成纤维细胞而非心肌成肌细胞的 分泌。功能丧失或获得研究表明,KLF15通过其反式激活结构域(TAD)负向调节 表达。有趣的是,腺病毒介导的全长KLF15——而非TAD缺失的KLF15过表达——显著预防了输注Ang II小鼠的病理变化。值得注意的是,给予 抑制剂SB265610可逆转KLF15基因敲除介导的由Ang II诱导的心脏功能障碍、重塑和炎症的加重。总之,我们的研究表明,心脏成纤维细胞中的KLF15负向调节 轴介导的炎症反应以及高血压中的后续心脏重塑。