Vanga Sudarsana Reddy, Åqvist Johan, Hallberg Anders, Gutiérrez-de-Terán Hugo
Department of Cell and Molecular Biology, BMC, Uppsala University, Uppsala, Sweden.
Department of Pharmaceutical Chemistry, BMC, Uppsala University, Uppsala, Sweden.
Front Mol Biosci. 2021 Apr 1;8:625274. doi: 10.3389/fmolb.2021.625274. eCollection 2021.
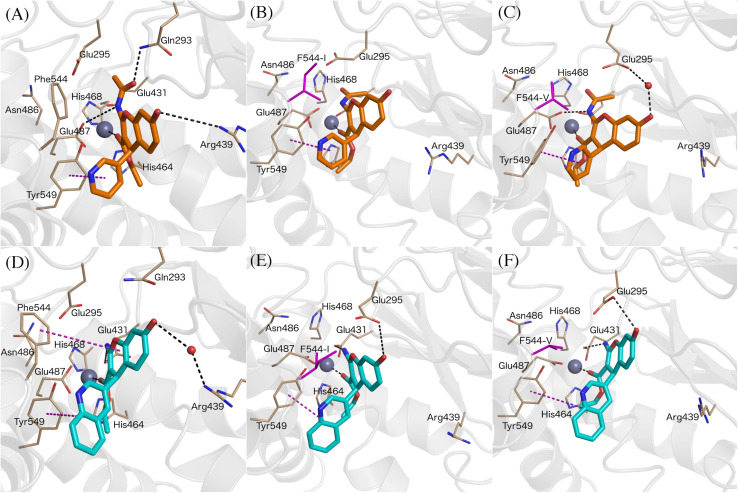
Inhibition of the insulin-regulated aminopeptidase (IRAP) improves memory and cognition in animal models. The enzyme has recently been crystallized and several series of inhibitors reported. We herein focused on one series of benzopyran-based inhibitors of IRAP known as the HFI series, with unresolved binding mode to IRAP, and developed a robust computational model to explain the structure-activity relationship (SAR) and potentially guide their further optimization. The binding model here proposed places the benzopyran ring in the catalytic binding site, coordinating the Zn ion through the oxygen in position 3, in contrast to previous hypothesis. The whole series of HFI compounds was then systematically simulated, starting from this binding mode, using molecular dynamics and binding affinity estimated with the linear interaction energy (LIE) method. The agreement with experimental affinities supports the binding mode proposed, which was further challenged by rigorous free energy perturbation (FEP) calculations. Here, we found excellent correlation between experimental and calculated binding affinity differences, both between selected compound pairs and also for recently reported experimental data concerning the site directed mutagenesis of residue Phe544. The computationally derived structure-activity relationship of the HFI series and the understanding of the involvement of Phe544 in the binding of this scaffold provide valuable information for further lead optimization of novel IRAP inhibitors.
抑制胰岛素调节氨肽酶(IRAP)可改善动物模型的记忆和认知能力。该酶最近已结晶,并报道了几个系列的抑制剂。我们在此聚焦于一系列基于苯并吡喃的IRAP抑制剂,即HFI系列,其与IRAP的结合模式尚未明确,我们开发了一个强大的计算模型来解释构效关系(SAR),并可能指导其进一步优化。与先前的假设相反,这里提出的结合模型将苯并吡喃环置于催化结合位点,通过3位的氧与锌离子配位。然后从这种结合模式开始,使用分子动力学和用线性相互作用能(LIE)方法估计的结合亲和力,对整个HFI化合物系列进行系统模拟。与实验亲和力的一致性支持了所提出的结合模式,而严格的自由能扰动(FEP)计算对其提出了进一步挑战。在这里,我们发现所选化合物对之间以及最近报道的关于残基Phe544定点诱变的实验数据的实验和计算结合亲和力差异之间具有极好的相关性。HFI系列的计算得出的构效关系以及对Phe544参与该支架结合的理解为新型IRAP抑制剂的进一步先导优化提供了有价值的信息。