Institute of Neurosciences and Department of Neurology of The Second Affiliated Hospital of Guangzhou Medical University and Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, 250 Changgang Dong RD, Guangzhou, 510260, People's Republic of China.
J Neuroinflammation. 2021 Apr 20;18(1):97. doi: 10.1186/s12974-021-02141-y.
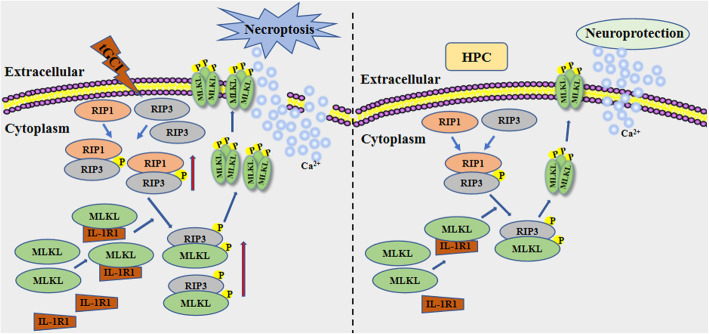
Our previous study indicated that hypoxic preconditioning reduced receptor interacting protein (RIP) 3-mediated necroptotic neuronal death in hippocampal CA1 of adult rats after transient global cerebral ischemia (tGCI). Although mixed lineage kinase domain-like (MLKL) has emerged as a crucial molecule for necroptosis induction downstream of RIP3, how MLKL executes necroptosis is not yet well understood. In this study, we aim to elucidate the molecular mechanism underlying hypoxic preconditioning that inactivates MLKL-dependent neuronal necroptosis after tGCI.
Transient global cerebral ischemia was induced by the four-vessel occlusion method. Twenty-four hours before ischemia, rats were exposed to systemic hypoxia with 8% O for 30 min. Western blotting was used to detect the expression of MLKL and interleukin-1 type 1 receptor (IL-1R1) in CA1. Immunoprecipitation was used to assess the interactions among IL-1R1, RIP3, and phosphorylated MLKL (p-MLKL). The concentration of intracellular free calcium ion (Ca) was measured using Fluo-4 AM. Silencing and overexpression studies were used to study the role of p-MLKL in tGCI-induced neuronal death.
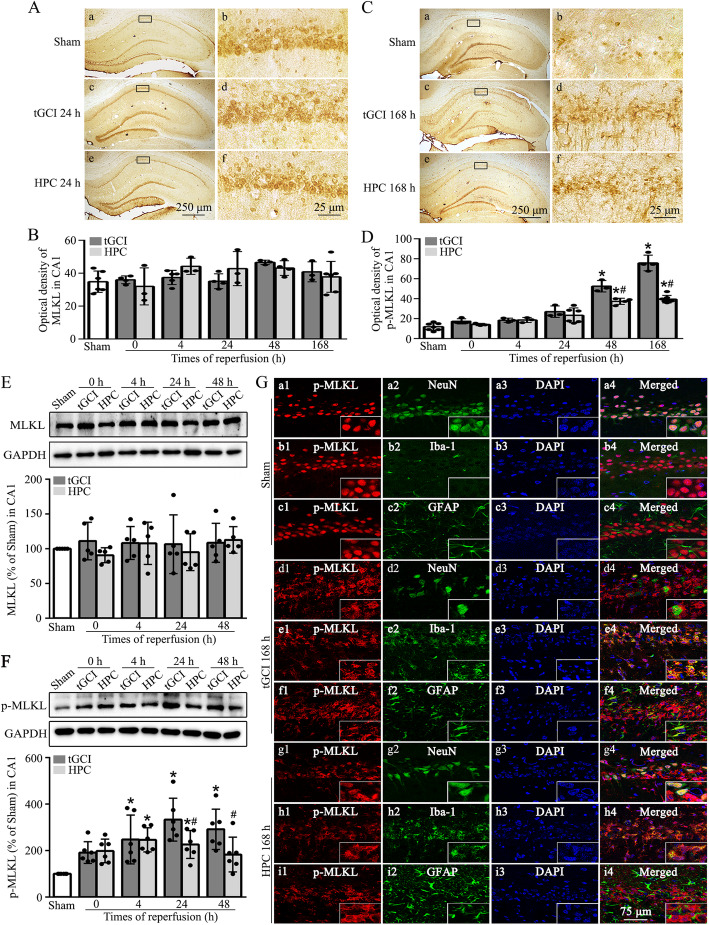
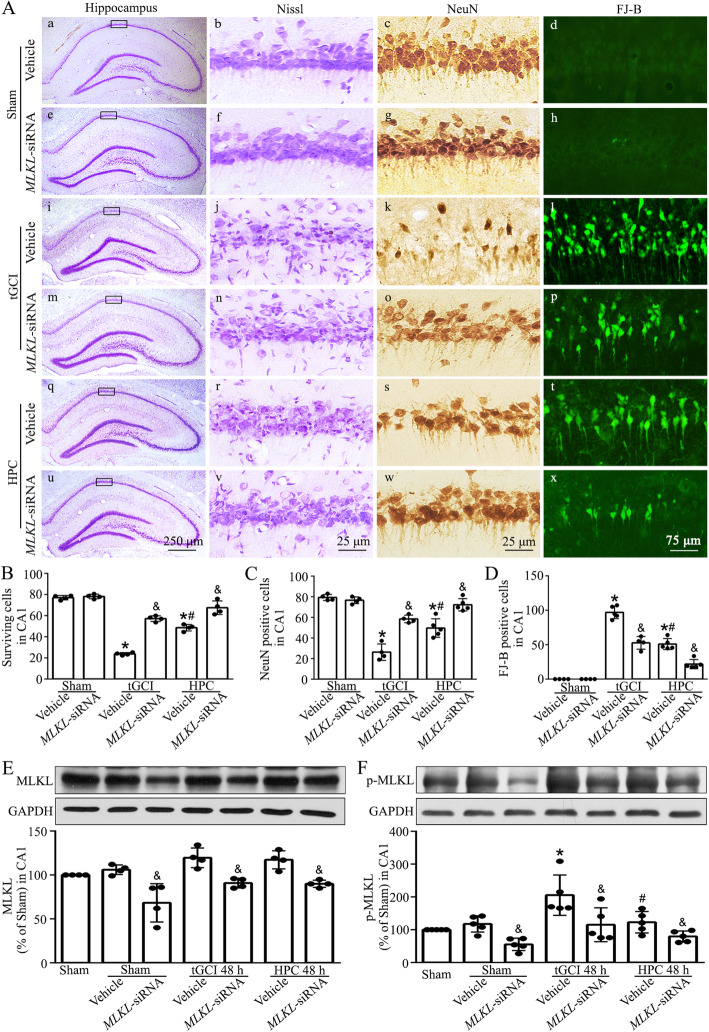
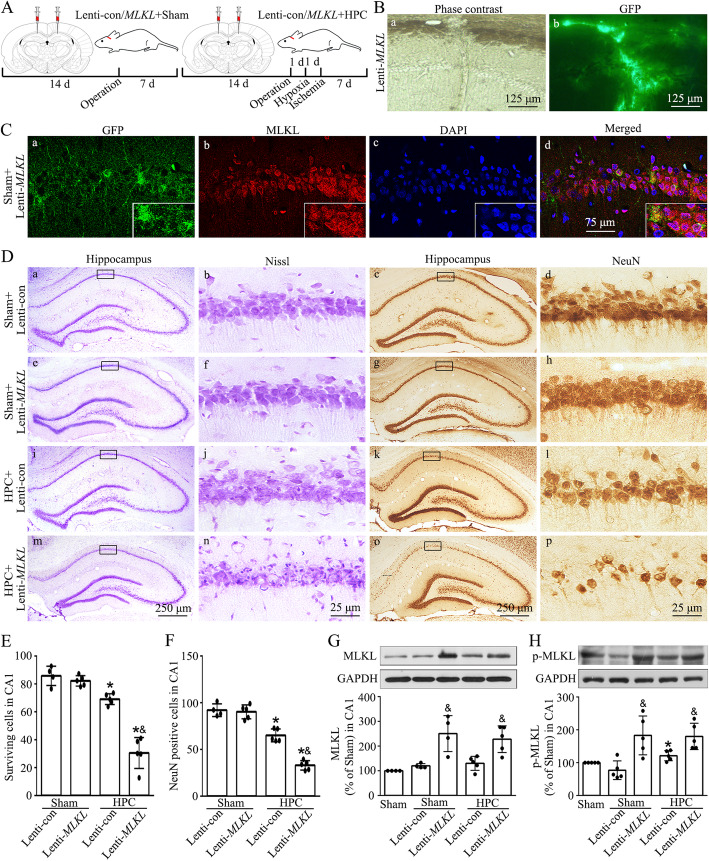
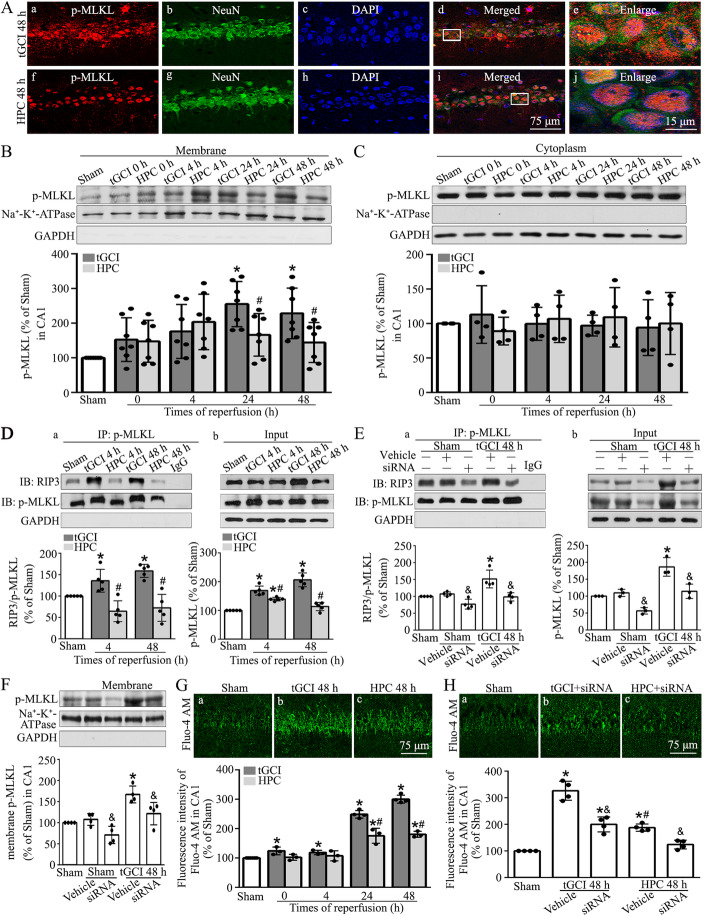
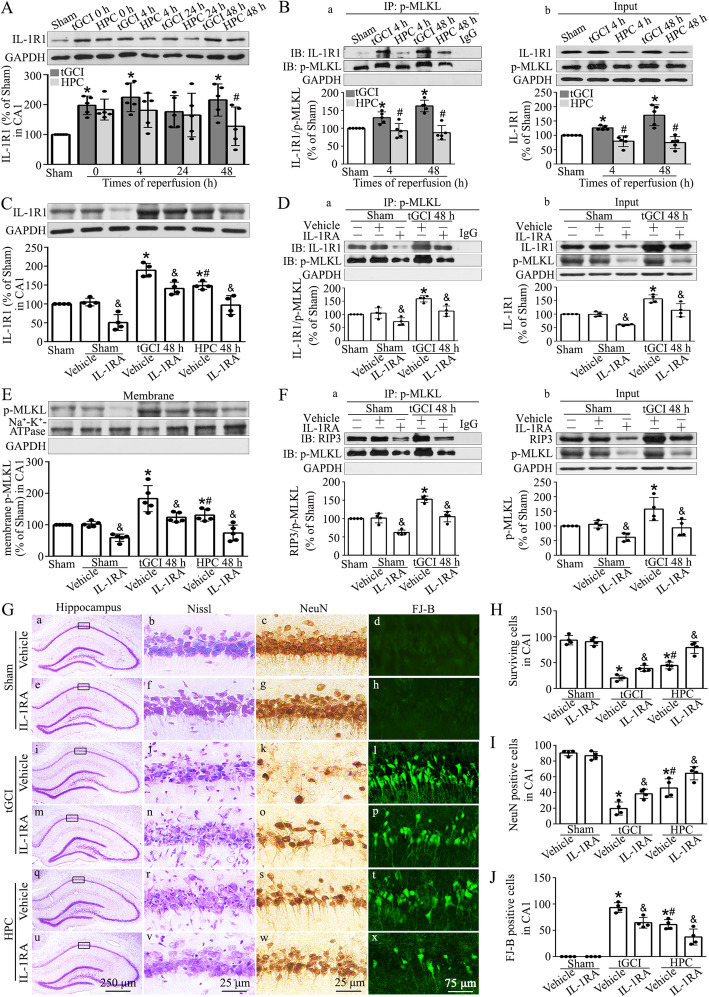
Hypoxic preconditioning decreased the phosphorylation of MLKL both in neurons and microglia of CA1 after tGCI. The knockdown of MLKL with siRNA decreased the expression of p-MLKL and exerted neuroprotective effects after tGCI, whereas treatment with lentiviral delivery of MLKL showed opposite results. Mechanistically, hypoxic preconditioning or MLKL siRNA attenuated the RIP3-p-MLKL interaction, reduced the plasma membrane translocation of p-MLKL, and blocked Ca influx after tGCI. Furthermore, hypoxic preconditioning downregulated the expression of IL-1R1 in CA1 after tGCI. Additionally, neutralizing IL-1R1 with its antagonist disrupted the interaction between IL-1R1 and the necrosome, attenuated the expression and the plasma membrane translocation of p-MLKL, thus alleviating neuronal death after tGCI.
These data support that the inhibition of MLKL-dependent neuronal necroptosis through downregulating IL-1R1 contributes to neuroprotection of hypoxic preconditioning against tGCI.
我们之前的研究表明,缺氧预处理可减少成年大鼠短暂全脑缺血(tGCI)后海马 CA1 中受体相互作用蛋白(RIP)3 介导的坏死性神经元死亡。虽然混合谱系激酶结构域样(MLKL)已成为 RIP3 下游诱导坏死性细胞死亡的关键分子,但 MLKL 如何执行坏死性细胞死亡尚不清楚。在这项研究中,我们旨在阐明缺氧预处理的分子机制,该机制可使 tGCI 后依赖 MLKL 的神经元坏死性细胞死亡失活。
通过四血管闭塞法诱导短暂性全脑缺血。在缺血前 24 小时,用 8%O2 对大鼠进行全身缺氧处理 30 分钟。Western blot 用于检测 CA1 中 MLKL 和白细胞介素 1 型 1 型受体(IL-1R1)的表达。免疫沉淀用于评估 IL-1R1、RIP3 和磷酸化 MLKL(p-MLKL)之间的相互作用。使用 Fluo-4 AM 测量细胞内游离钙离子(Ca)浓度。沉默和过表达研究用于研究 p-MLKL 在 tGCI 诱导的神经元死亡中的作用。
缺氧预处理可降低 tGCI 后 CA1 神经元和小胶质细胞中 MLKL 的磷酸化。用 siRNA 沉默 MLKL 可降低 p-MLKL 的表达并发挥 tGCI 后的神经保护作用,而用慢病毒转染 MLKL 则产生相反的结果。机制上,缺氧预处理或 MLKL siRNA 减弱了 RIP3-p-MLKL 相互作用,减少了 p-MLKL 的质膜易位,并阻断了 tGCI 后的 Ca 内流。此外,缺氧预处理可降低 tGCI 后 CA1 中 IL-1R1 的表达。此外,用其拮抗剂中和 IL-1R1 可破坏 IL-1R1 和坏死小体之间的相互作用,减弱 p-MLKL 的表达和质膜易位,从而减轻 tGCI 后的神经元死亡。
这些数据支持通过下调 IL-1R1 抑制依赖 MLKL 的神经元坏死性细胞死亡有助于缺氧预处理对 tGCI 的神经保护作用。