Institute of Electronics, Computer and Telecommunication Engineering, National Research Council of Italy, Genoa, Italy.
Computer Laboratory, University of Cambridge, Cambridge, UK.
BMC Bioinformatics. 2021 Apr 26;22(Suppl 2):43. doi: 10.1186/s12859-020-03937-0.
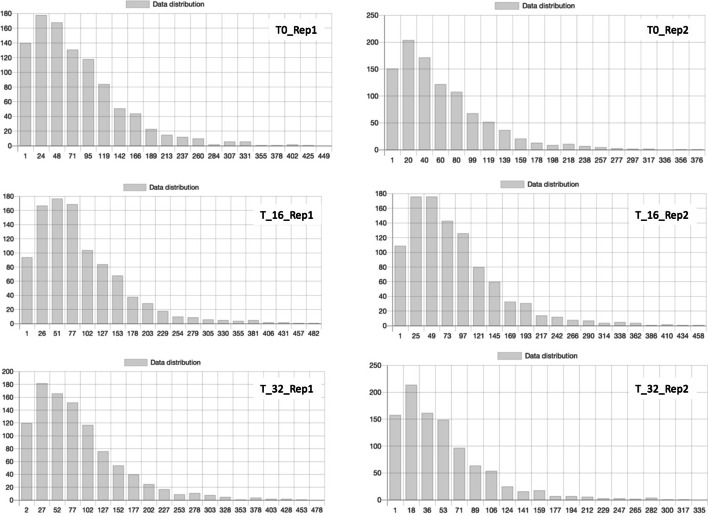
High-throughput sequencing Chromosome Conformation Capture (Hi-C) allows the study of DNA interactions and 3D chromosome folding at the genome-wide scale. Usually, these data are represented as matrices describing the binary contacts among the different chromosome regions. On the other hand, a graph-based representation can be advantageous to describe the complex topology achieved by the DNA in the nucleus of eukaryotic cells.
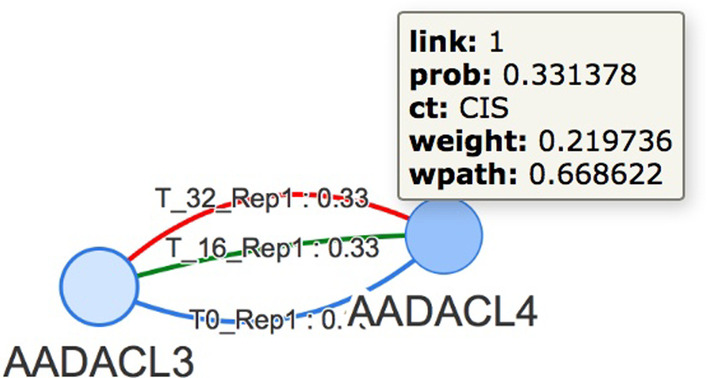
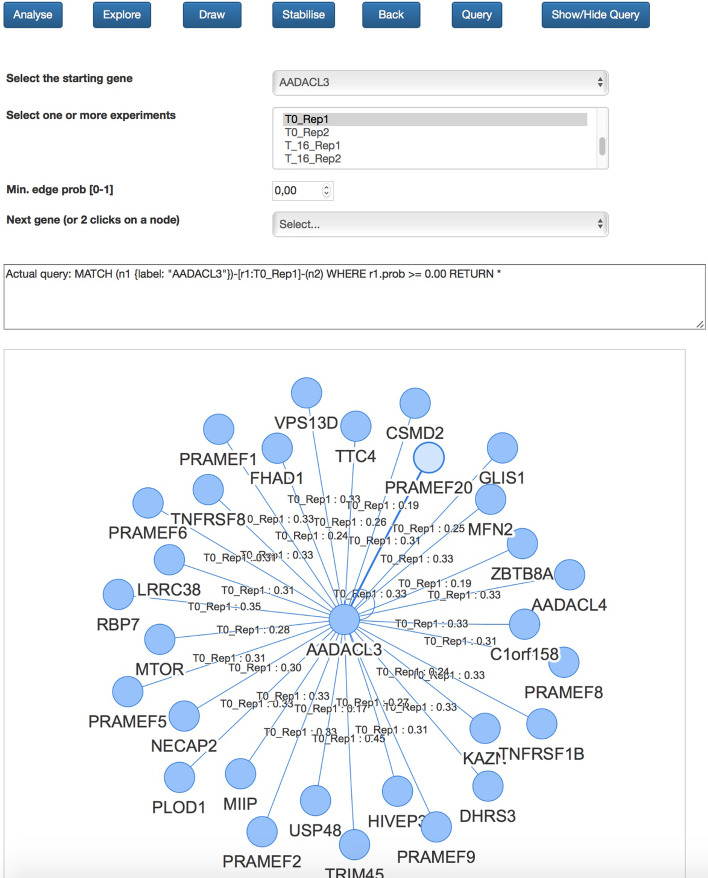
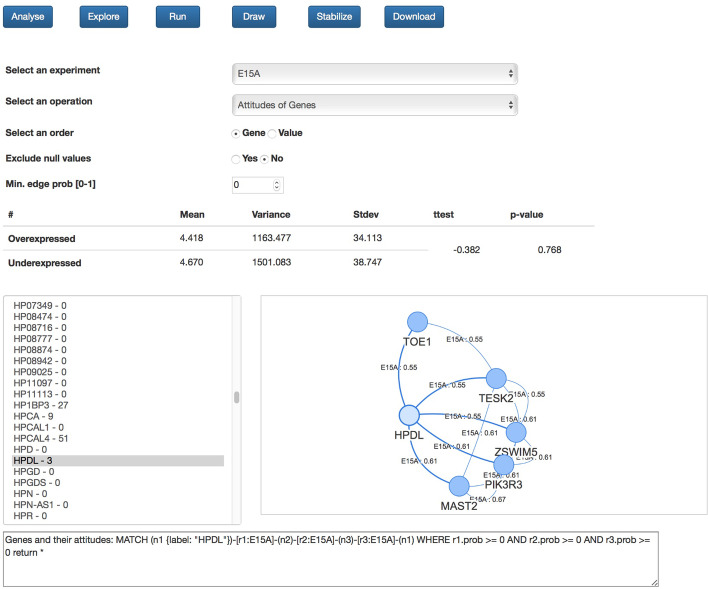
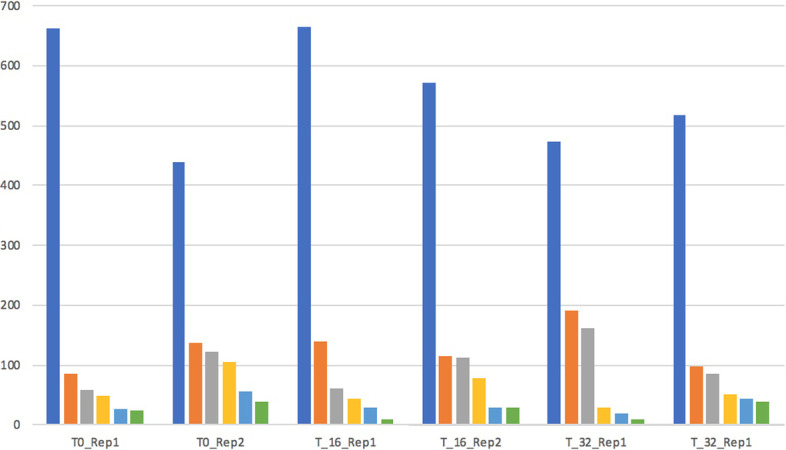
Here we discuss the use of a graph database for storing and analysing data achieved by performing Hi-C experiments. The main issue is the size of the produced data and, working with a graph-based representation, the consequent necessity of adequately managing a large number of edges (contacts) connecting nodes (genes), which represents the sources of information. For this, currently available graph visualisation tools and libraries fall short with Hi-C data. The use of graph databases, instead, supports both the analysis and the visualisation of the spatial pattern present in Hi-C data, in particular for comparing different experiments or for re-mapping omics data in a space-aware context efficiently. In particular, the possibility of describing graphs through statistical indicators and, even more, the capability of correlating them through statistical distributions allows highlighting similarities and differences among different Hi-C experiments, in different cell conditions or different cell types.
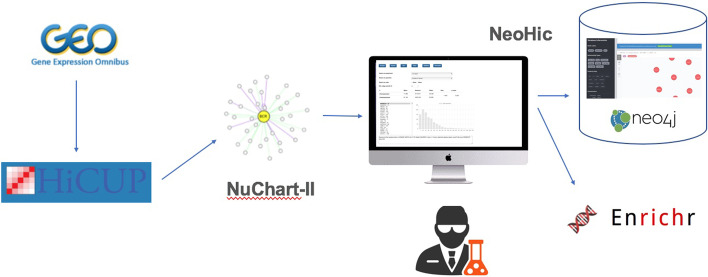
These concepts have been implemented in NeoHiC, an open-source and user-friendly web application for the progressive visualisation and analysis of Hi-C networks based on the use of the Neo4j graph database (version 3.5).
With the accumulation of more experiments, the tool will provide invaluable support to compare neighbours of genes across experiments and conditions, helping in highlighting changes in functional domains and identifying new co-organised genomic compartments.
高通量测序染色体构象捕获(Hi-C)允许在全基因组范围内研究 DNA 相互作用和 3D 染色体折叠。通常,这些数据表示为描述不同染色体区域之间二进制接触的矩阵。另一方面,基于图的表示形式有利于描述真核细胞核中 DNA 所实现的复杂拓扑结构。
在这里,我们讨论了使用图形数据库存储和分析通过执行 Hi-C 实验获得的数据。主要问题是产生的数据的大小,并且,使用基于图的表示形式,需要适当地管理连接节点(基因)的大量边(接触),这是信息的来源。为此,目前可用的图形可视化工具和库在处理 Hi-C 数据方面存在不足。相反,图形数据库的使用支持对 Hi-C 数据中存在的空间模式进行分析和可视化,特别是用于有效地比较不同的实验或在空间感知上下文中重新映射组学数据。特别是,通过统计指标来描述图的可能性,甚至更重要的是,通过统计分布来关联它们的能力,可以突出不同 Hi-C 实验、不同细胞条件或不同细胞类型之间的相似性和差异。
这些概念已在 NeoHiC 中实现,这是一种开源且用户友好的网络应用程序,用于基于 Neo4j 图形数据库(版本 3.5)渐进式可视化和分析 Hi-C 网络。
随着更多实验的积累,该工具将为比较跨实验和条件的基因邻居提供宝贵的支持,帮助突出功能域的变化并识别新的组织基因组区室。