Department of Physiology, School of Medicine, Fukuoka University, Fukuoka 814-0180, Japan.
Biomedical Information Engineering Lab, The University of Aizu, Aizu-Wakamatsu 965-8580, Japan.
Cells. 2021 Apr 22;10(5):983. doi: 10.3390/cells10050983.
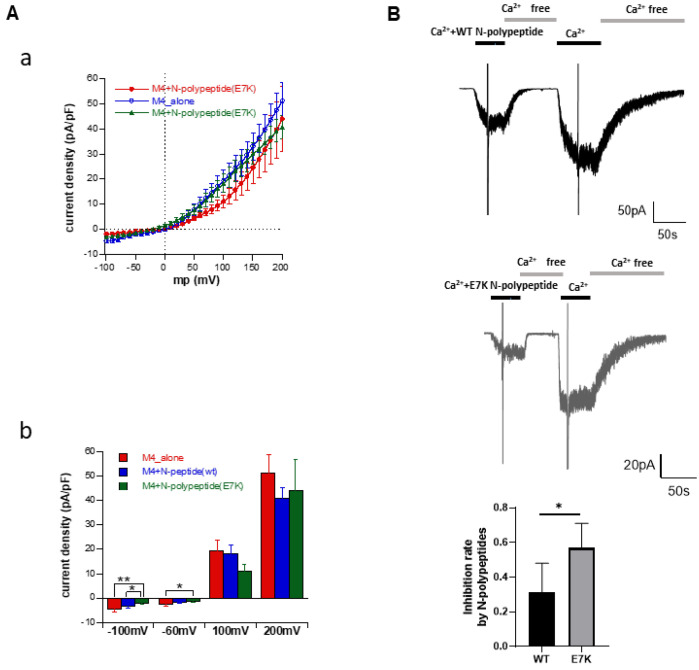
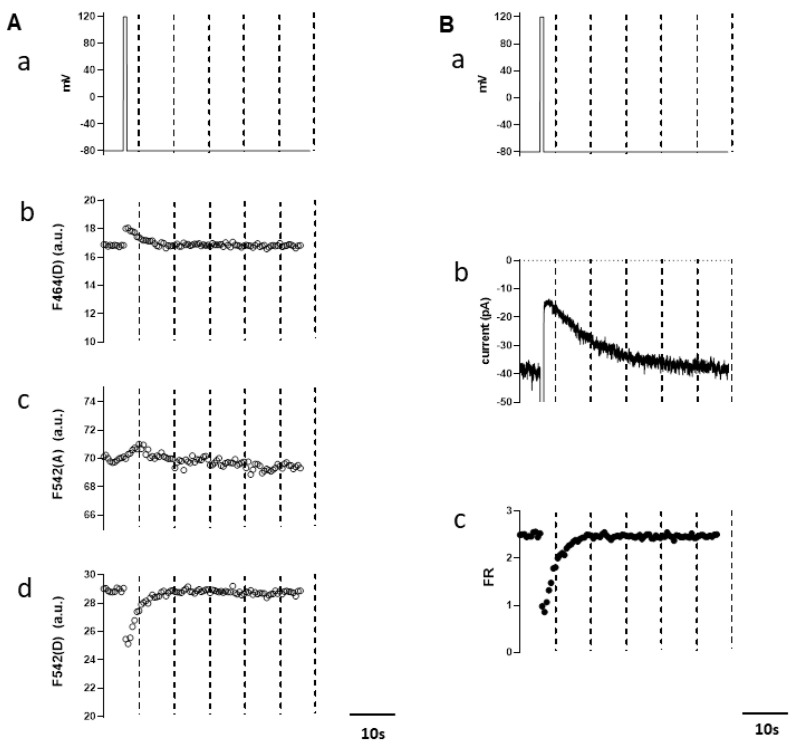
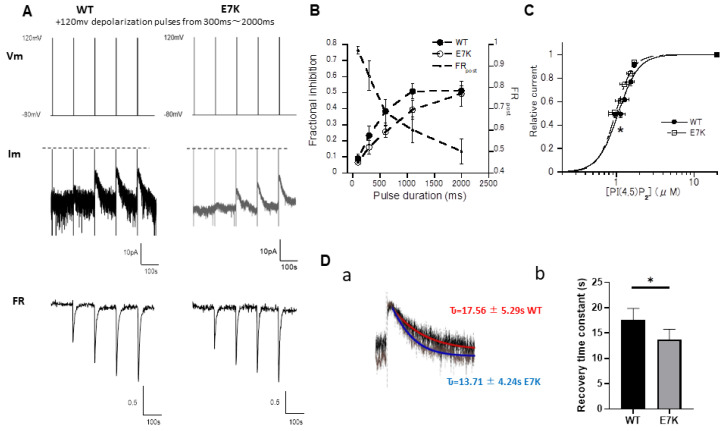
A Ca-activated monovalent cation-selective TRPM4 channel is abundantly expressed in the heart. Recently, a single gain-of-function mutation identified in the distal N-terminus of the human TRPM4 channel (Glu to Lys; E7K) was found to be arrhythmogenic because of enhanced cell membrane expression. In this study, we conducted detailed analyses of this mutant channel from more functional aspects, in comparison with its wild type (WT). In an expression system, intracellular application of a short soluble PIP (diCPIP) restored the single-channel activities of both WT and E7K, which had quickly faded after membrane excision. The potency (K) of diCPIP for this recovery was stronger in E7K than its WT (1.44 vs. 2.40 μM). FRET-based PIP measurements combined with the voltage-sensing phosphatase (DrVSP) and patch clamping revealed that lowering the endogenous PIP level by DrVSP activation reduced the TRPM4 channel activity. This effect was less prominent in E7K than its WT (apparent K values estimated from DrVSP-mediated PIP depletion: 0.97 and 1.06 μM, respectively), being associated with the differential PIP-mediated modulation of voltage dependence. Moreover, intracellular perfusion of short N-terminal polypeptides containing either the 'WT' or 'E7K' sequences respectively attenuated the TRPM4 channel activation at whole-cell and single-channel levels, but in both configurations, the E7K polypeptide exerted greater inhibitory effects. These results collectively suggest that N-terminal interaction with endogenous PIP is essential for the TRPM4 channel to function, the extent of which may be abnormally strengthened by the E7K mutation through modulating voltage-dependent activation. The altered PIP interaction may account for the arrhythmogenic potential of this mutation.
一种钙激活的单价阳离子选择性 TRPM4 通道在心脏中大量表达。最近,在人类 TRPM4 通道的远端 N 端发现了一个单一的功能获得性突变(Glu 突变为 Lys;E7K),由于增强了细胞膜表达,被认为是致心律失常的。在这项研究中,我们从更多功能方面对这种突变通道进行了详细分析,与野生型(WT)进行了比较。在表达系统中,短可溶性 PIP(diCPIP)的细胞内应用恢复了 WT 和 E7K 的单通道活性,这两种活性在膜切除后很快消失。diCPIP 对这种恢复的效力在 E7K 中比其 WT 更强(1.44 对 2.40 μM)。基于 FRET 的 PIP 测量与电压感应磷酸酶(DrVSP)和膜片钳结合显示,通过 DrVSP 激活降低内源性 PIP 水平会降低 TRPM4 通道活性。这种效应在 E7K 中不如其 WT 明显(从 DrVSP 介导的 PIP 耗竭估计的表观 K 值:分别为 0.97 和 1.06 μM),这与电压依赖性调制的差异相关。此外,含有“WT”或“E7K”序列的短 N 端多肽的细胞内灌注分别减弱了全细胞和单通道水平的 TRPM4 通道激活,但在这两种构型中,E7K 多肽均发挥了更大的抑制作用。这些结果共同表明,N 端与内源性 PIP 的相互作用对于 TRPM4 通道的功能至关重要,E7K 突变可能通过调节电压依赖性激活来异常增强这种相互作用。改变的 PIP 相互作用可能是这种突变致心律失常潜力的原因。