Bludau Isabell
Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, 82152 Martinsried, Germany.
Int J Mol Sci. 2021 Apr 24;22(9):4450. doi: 10.3390/ijms22094450.
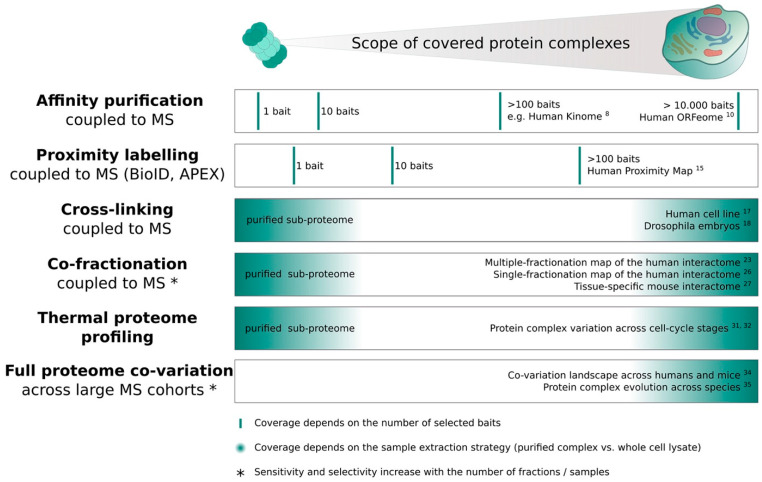
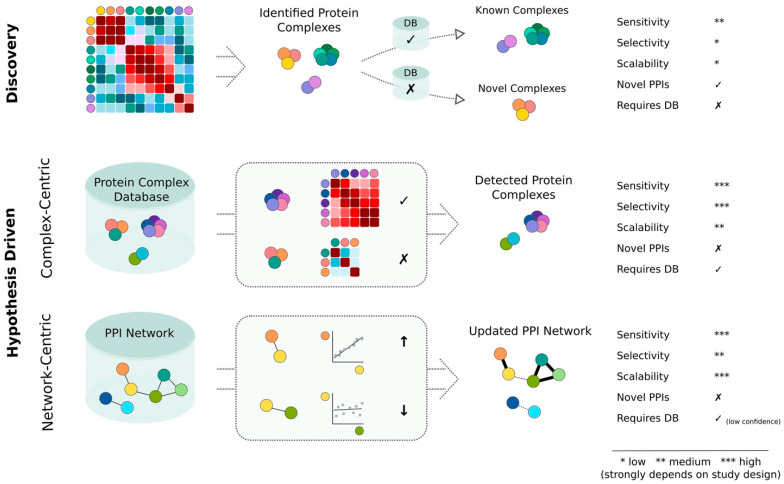
Protein complexes are the main functional modules in the cell that coordinate and perform the vast majority of molecular functions. The main approaches to identify and quantify the interactome to date are based on mass spectrometry (MS). Here I summarize the benefits and limitations of different MS-based interactome screens, with a focus on untargeted interactome acquisition, such as co-fractionation MS. Specific emphasis is given to the discussion of discovery- versus hypothesis-driven data analysis concepts and their applicability to large, proteome-wide interactome screens. Hypothesis-driven analysis approaches, i.e., complex- or network-centric, are highlighted as promising strategies for comparative studies. While these approaches require prior information from public databases, also reviewed herein, the available wealth of interactomic data continuously increases, thereby providing more exhaustive information for future studies. Finally, guidance on the selection of interactome acquisition and analysis methods is provided to aid the reader in the design of protein-protein interaction studies.
蛋白质复合物是细胞中的主要功能模块,它们协调并执行绝大多数分子功能。迄今为止,识别和量化相互作用组的主要方法基于质谱(MS)。在此,我总结了不同基于质谱的相互作用组筛选方法的优缺点,重点关注非靶向相互作用组获取,如共分离质谱。特别强调了对发现驱动与假设驱动数据分析概念及其在大规模、全蛋白质组相互作用组筛选中的适用性的讨论。假设驱动的分析方法,即以复合物或网络为中心的方法,被突出为比较研究的有前景策略。虽然这些方法需要来自公共数据库的先验信息(本文也对此进行了综述),但可用的相互作用组数据量在不断增加,从而为未来研究提供了更详尽的信息。最后,提供了相互作用组获取和分析方法选择的指导,以帮助读者设计蛋白质 - 蛋白质相互作用研究。