Department of Molecular Biosciences, Center for Systems and Synthetic Biology, University of Texas, Austin, TX, USA.
Mol Syst Biol. 2021 May;17(5):e10016. doi: 10.15252/msb.202010016.
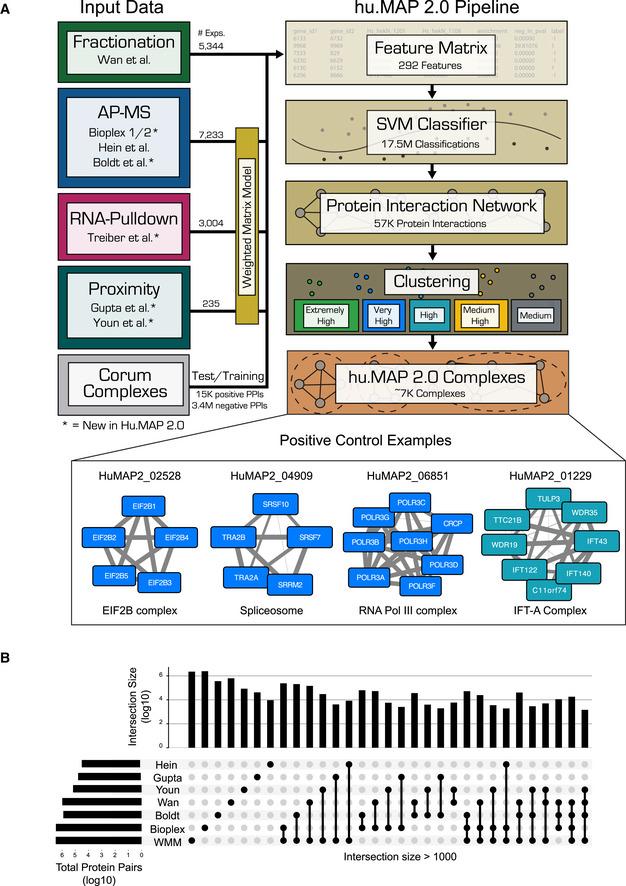
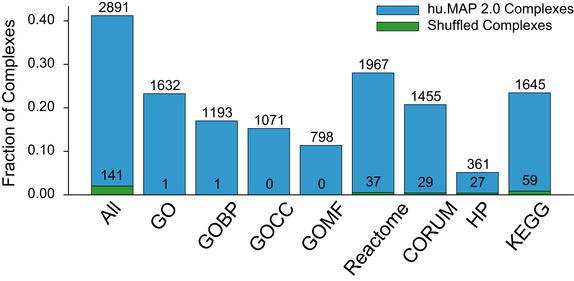
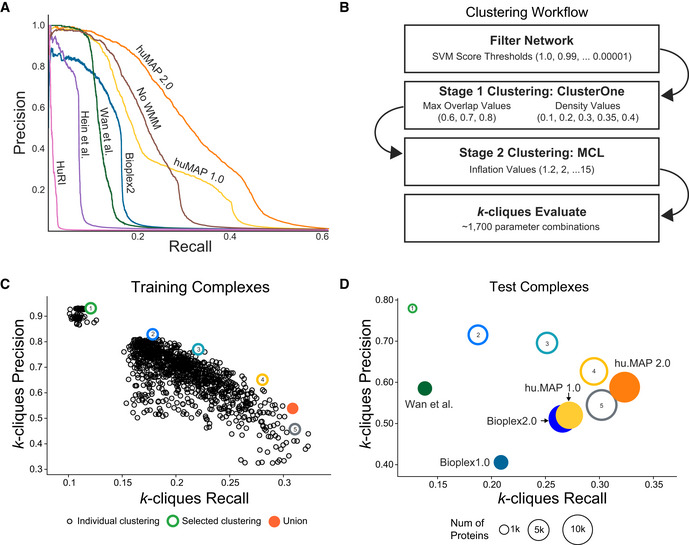
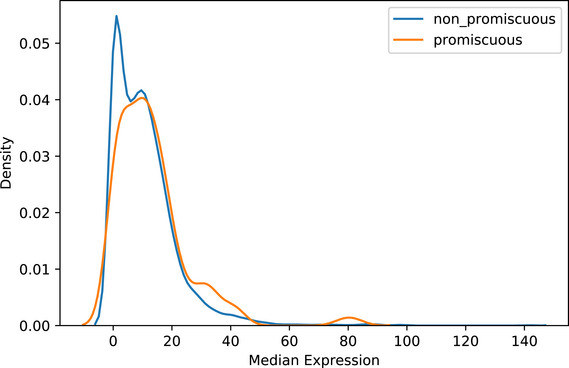
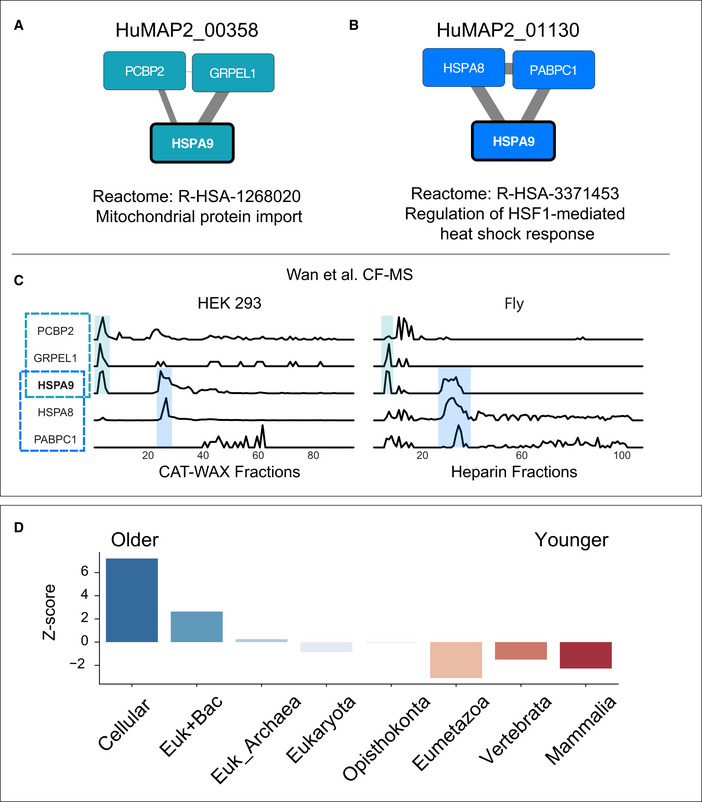
A general principle of biology is the self-assembly of proteins into functional complexes. Characterizing their composition is, therefore, required for our understanding of cellular functions. Unfortunately, we lack knowledge of the comprehensive set of identities of protein complexes in human cells. To address this gap, we developed a machine learning framework to identify protein complexes in over 15,000 mass spectrometry experiments which resulted in the identification of nearly 7,000 physical assemblies. We show our resource, hu.MAP 2.0, is more accurate and comprehensive than previous state of the art high-throughput protein complex resources and gives rise to many new hypotheses, including for 274 completely uncharacterized proteins. Further, we identify 253 promiscuous proteins that participate in multiple complexes pointing to possible moonlighting roles. We have made hu.MAP 2.0 easily searchable in a web interface (http://humap2.proteincomplexes.org/), which will be a valuable resource for researchers across a broad range of interests including systems biology, structural biology, and molecular explanations of disease.
生物学的一个普遍原则是蛋白质自我组装成功能复合物。因此,要了解细胞功能,就必须确定它们的组成。遗憾的是,我们并不了解人类细胞中蛋白质复合物的全面组成。为了解决这一差距,我们开发了一种机器学习框架,用于鉴定超过 15000 次质谱实验中的蛋白质复合物,这导致了近 7000 个物理组装体的鉴定。我们的资源 hu.MAP 2.0 比以前的高通量蛋白质复合物资源更准确、更全面,并产生了许多新的假设,包括 274 个完全未知的蛋白质。此外,我们还鉴定出 253 个参与多个复合物的多功能蛋白,这可能指向潜在的兼职作用。我们已经在网络界面(http://humap2.proteincomplexes.org/)中使 hu.MAP 2.0 易于搜索,这将成为包括系统生物学、结构生物学和疾病分子解释在内的广泛研究领域的研究人员的宝贵资源。