Yu Wenbo, Mahfouz Ahmed, Reinders Marcel J T
Department of Control Science and Engineering, Harbin Institute of Technology, Harbin, China.
Delft Bioinformatics Lab, Delft University of Technology, Delft, Netherlands.
Front Genet. 2021 Apr 13;12:644211. doi: 10.3389/fgene.2021.644211. eCollection 2021.
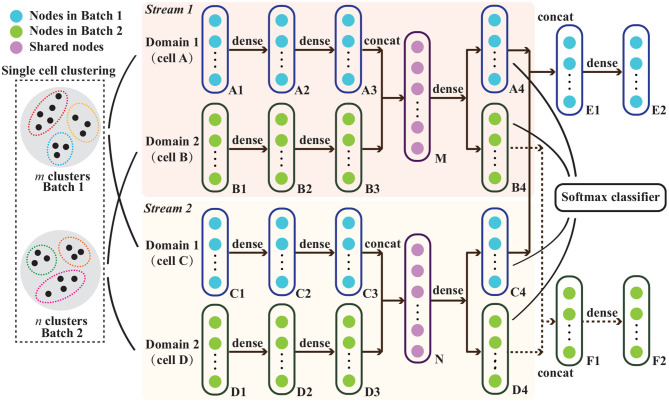
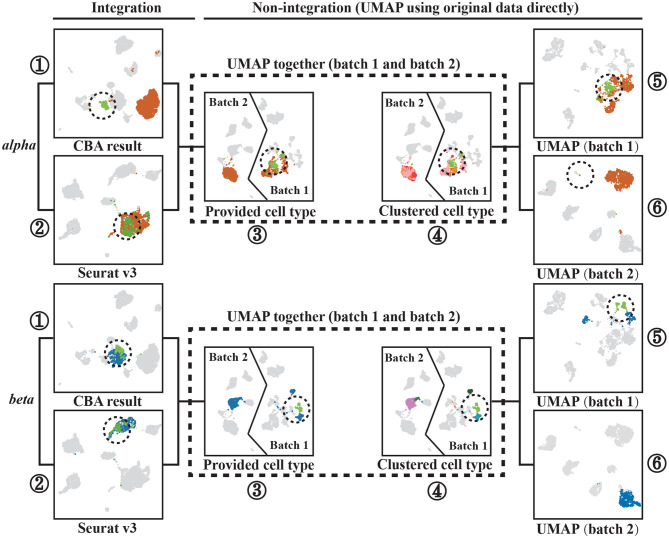
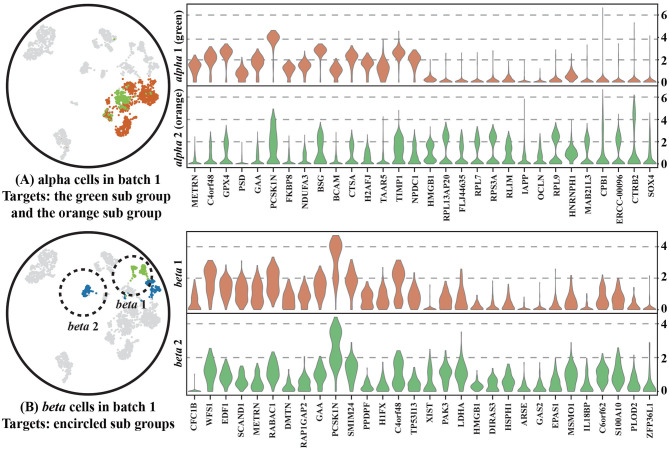
The power of single-cell RNA sequencing (scRNA-seq) in detecting cell heterogeneity or developmental process is becoming more and more evident every day. The granularity of this knowledge is further propelled when combining two batches of scRNA-seq into a single large dataset. This strategy is however hampered by technical differences between these batches. Typically, these batch effects are resolved by matching similar cells across the different batches. Current approaches, however, do not take into account that we can constrain this matching further as cells can also be matched on their cell type identity. We use an auto-encoder to embed two batches in the same space such that cells are matched. To accomplish this, we use a loss function that preserves: (1) cell-cell distances within each of the two batches, as well as (2) cell-cell distances between two batches when the cells are of the same cell-type. The cell-type guidance is unsupervised, i.e., a cell-type is defined as a cluster in the original batch. We evaluated the performance of our cluster-guided batch alignment (CBA) using pancreas and mouse cell atlas datasets, against six state-of-the-art single cell alignment methods: Seurat v3, BBKNN, Scanorama, Harmony, LIGER, and BERMUDA. Compared to other approaches, CBA preserves the cluster separation in the original datasets while still being able to align the two datasets. We confirm that this separation is biologically meaningful by identifying relevant differential expression of genes for these preserved clusters.
单细胞RNA测序(scRNA-seq)在检测细胞异质性或发育过程中的强大作用日益凸显。将两批scRNA-seq合并为一个大型数据集时,这种知识的精细程度会进一步提升。然而,这一策略受到批次间技术差异的阻碍。通常,这些批次效应通过跨不同批次匹配相似细胞来解决。然而,目前的方法没有考虑到,由于细胞也可以根据其细胞类型身份进行匹配,我们可以进一步限制这种匹配。我们使用自动编码器将两批数据嵌入到同一空间中,以便细胞进行匹配。为实现这一点,我们使用一种损失函数来保留:(1)两批数据中每一批内的细胞间距离,以及(2)当细胞属于同一细胞类型时两批数据之间的细胞间距离。细胞类型引导是无监督的,即细胞类型在原始批次中被定义为一个聚类。我们使用胰腺和小鼠细胞图谱数据集评估了我们的聚类引导批次对齐(CBA)方法的性能,并与六种最先进的单细胞对齐方法进行了比较:Seurat v3、BBKNN、Scanorama、Harmony、LIGER和BERMUDA。与其他方法相比,CBA在保留原始数据集中聚类分离的同时,仍能够对齐两个数据集。我们通过识别这些保留聚类的相关基因差异表达来确认这种分离具有生物学意义。