Mandric Igor, Hill Brian L, Freund Malika K, Thompson Michael, Halperin Eran
Department of Computer Science, University of California Los Angeles, Los Angeles, CA, USA.
Department of Computer Science, University of California Los Angeles, Los Angeles, CA, USA.
iScience. 2020 Jun 26;23(6):101185. doi: 10.1016/j.isci.2020.101185. Epub 2020 May 20.
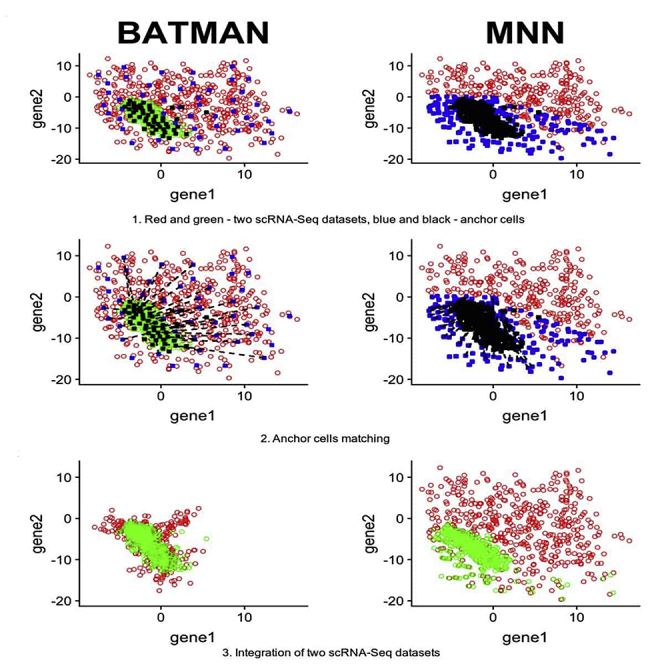
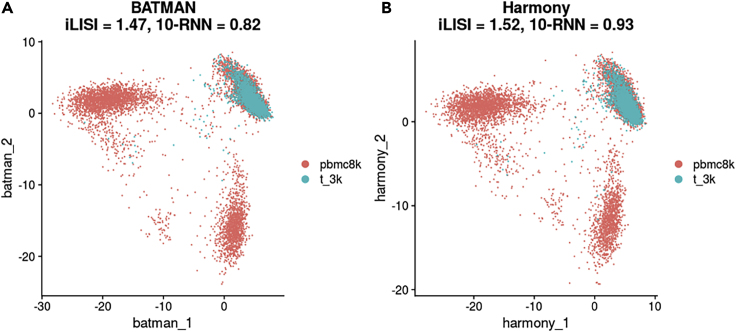
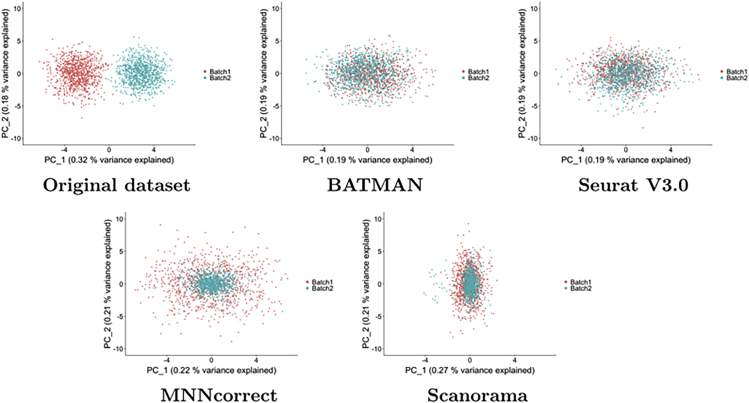
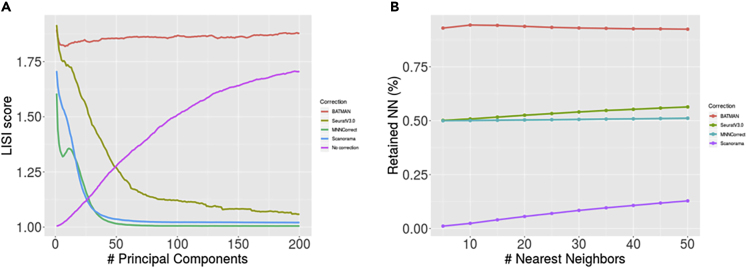
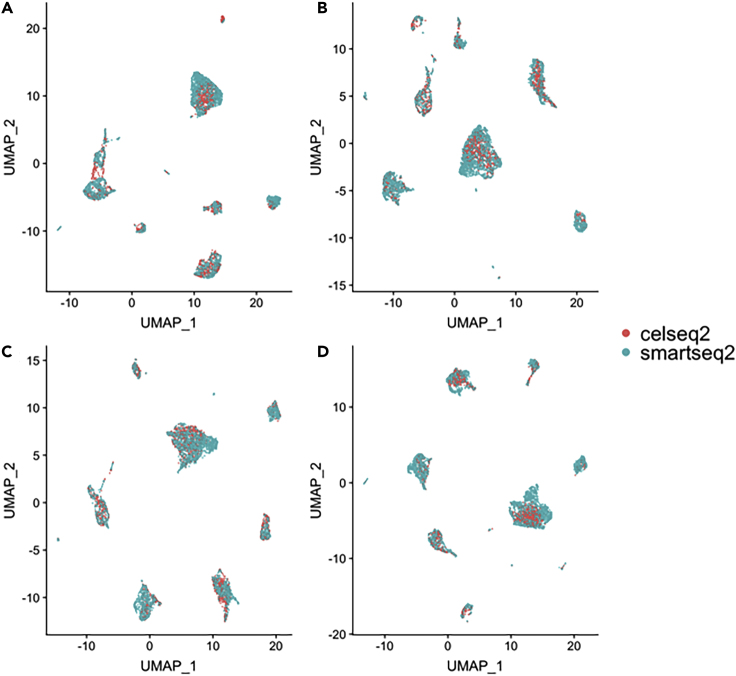
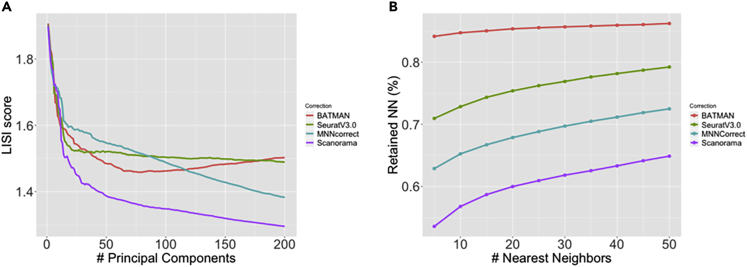
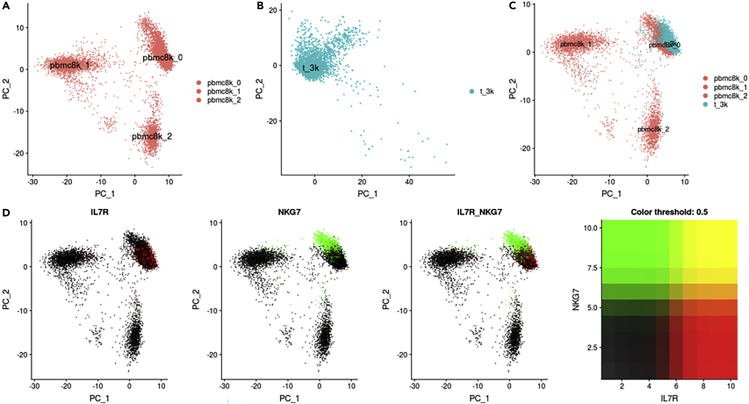
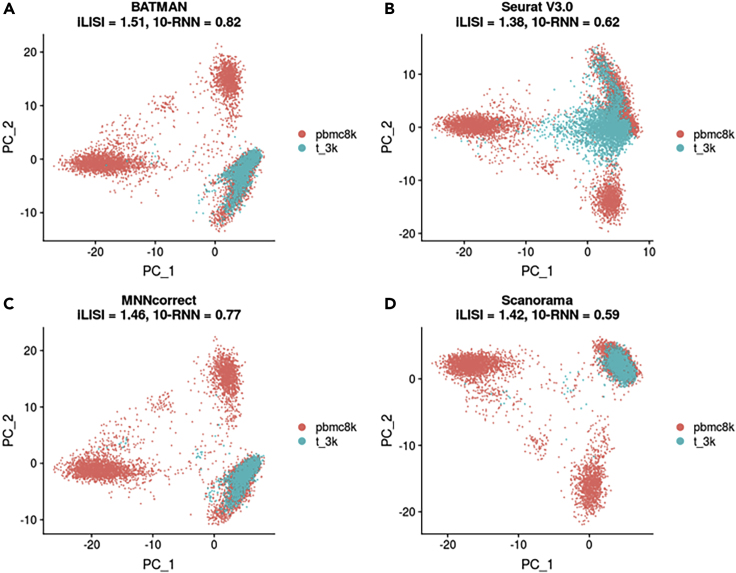
Single-cell RNA-sequencing (scRNA-seq) is a set of technologies used to profile gene expression at the level of individual cells. Although the throughput of scRNA-seq experiments is steadily growing in terms of the number of cells, large datasets are not yet commonly generated owing to prohibitively high costs. Integrating multiple datasets into one can improve power in scRNA-seq experiments, and efficient integration is very important for downstream analyses such as identifying cell-type-specific eQTLs. State-of-the-art scRNA-seq integration methods are based on the mutual nearest neighbor paradigm and fail to both correct for batch effects and maintain the local structure of the datasets. In this paper, we propose a novel scRNA-seq dataset integration method called BATMAN (BATch integration via minimum-weight MAtchiNg). Across multiple simulations and real datasets, we show that our method significantly outperforms state-of-the-art tools with respect to existing metrics for batch effects by up to 80% while retaining cell-to-cell relationships.
单细胞RNA测序(scRNA-seq)是一组用于在单个细胞水平上分析基因表达的技术。尽管scRNA-seq实验的通量在细胞数量方面稳步增长,但由于成本过高,尚未普遍生成大型数据集。将多个数据集整合为一个可以提高scRNA-seq实验的效能,而高效整合对于诸如识别细胞类型特异性eQTL等下游分析非常重要。当前最先进的scRNA-seq整合方法基于相互最近邻范式,无法同时校正批次效应并保持数据集的局部结构。在本文中,我们提出了一种名为BATMAN(通过最小权重匹配进行批次整合)的新型scRNA-seq数据集整合方法。在多个模拟和真实数据集上,我们表明,相对于现有的批次效应指标,我们的方法显著优于当前最先进的工具,最高可达80%,同时保留了细胞间关系。