Hiatt Susan M, Lawlor James M J, Handley Lori H, Ramaker Ryne C, Rogers Brianne B, Partridge E Christopher, Boston Lori Beth, Williams Melissa, Plott Christopher B, Jenkins Jerry, Gray David E, Holt James M, Bowling Kevin M, Bebin E Martina, Grimwood Jane, Schmutz Jeremy, Cooper Gregory M
HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806, USA.
Department of Genetics, University of Alabama at Birmingham, Birmingham, AL 35924, USA.
HGG Adv. 2021 Apr 8;2(2). doi: 10.1016/j.xhgg.2021.100023. Epub 2021 Jan 16.
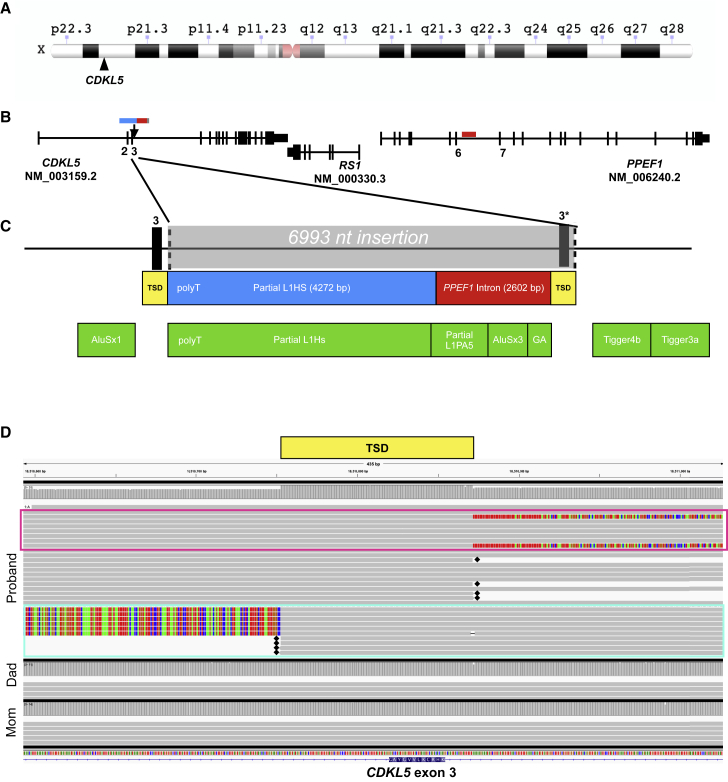
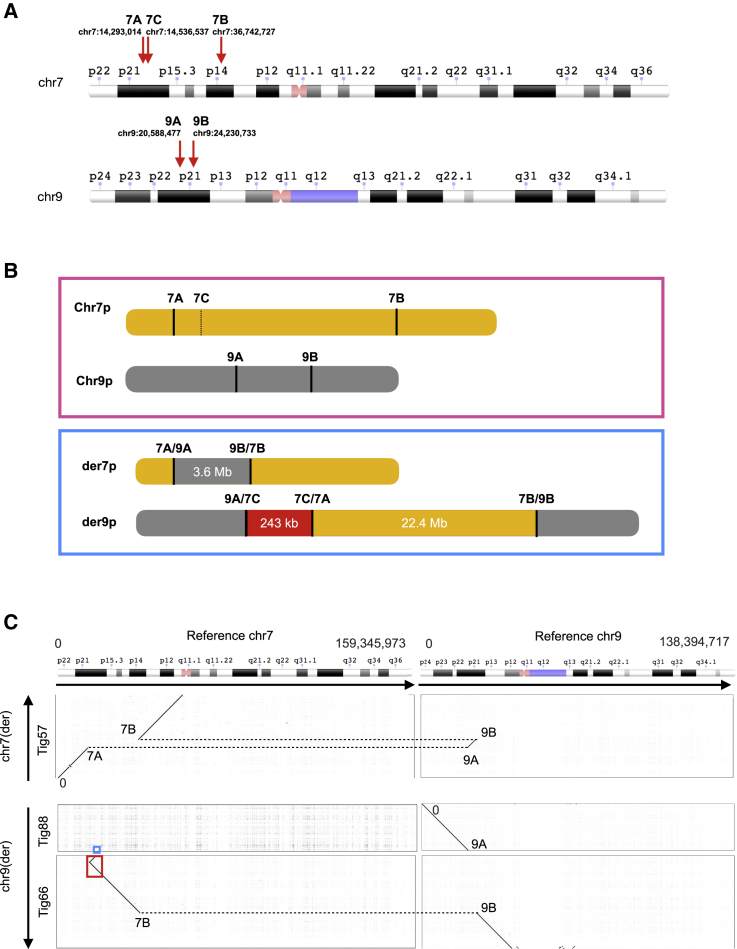
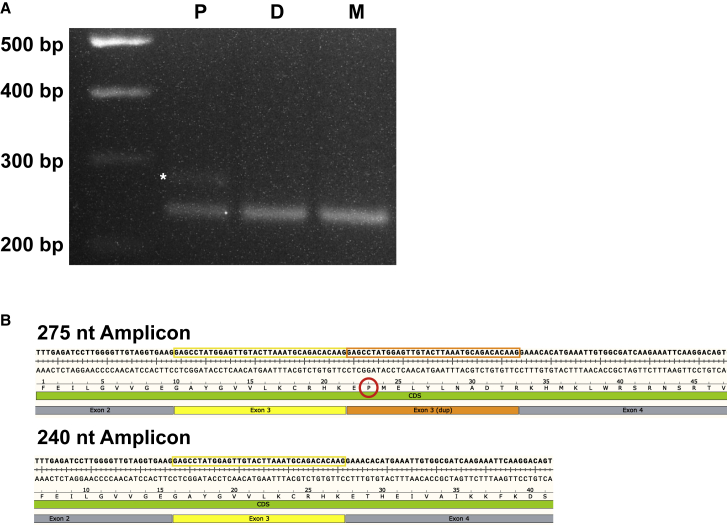
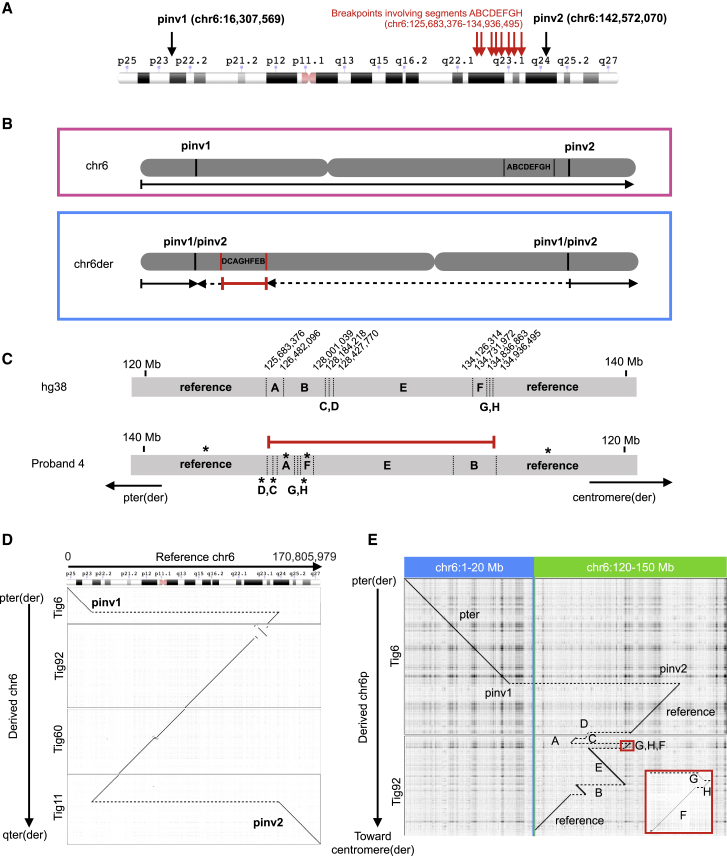
Exome and genome sequencing have proven to be effective tools for the diagnosis of neurodevelopmental disorders (NDDs), but large fractions of NDDs cannot be attributed to currently detectable genetic variation. This is likely, at least in part, a result of the fact that many genetic variants are difficult or impossible to detect through typical short-read sequencing approaches. Here, we describe a genomic analysis using Pacific Biosciences circular consensus sequencing (CCS) reads, which are both long (>10 kb) and accurate (>99% bp accuracy). We used CCS on six proband-parent trios with NDDs that were unexplained despite extensive testing, including genome sequencing with short reads. We identified variants and created assemblies in each trio, with global metrics indicating these datasets are more accurate and comprehensive than those provided by short-read data. In one proband, we identified a likely pathogenic (LP), L1-mediated insertion in that results in duplication of exon 3, leading to a frameshift. In a second proband, we identified multiple large structural variants, including insertion-translocations affecting and , which we show disrupt transcript levels. We consider this extensive structural variation likely pathogenic. The breadth and quality of variant detection, coupled to finding variants of clinical and research interest in two of six probands with unexplained NDDs, support the hypothesis that long-read genome sequencing can substantially improve rare disease genetic discovery rates.
外显子组测序和基因组测序已被证明是诊断神经发育障碍(NDDs)的有效工具,但很大一部分NDDs无法归因于目前可检测到的基因变异。这至少在部分程度上可能是由于许多基因变异难以或无法通过典型的短读长测序方法检测到。在这里,我们描述了一项使用太平洋生物科学公司环形一致序列测序(CCS)读数的基因组分析,这些读数既长(>10 kb)又准确(>99%碱基准确率)。我们对六个患有NDDs的先证者-父母三联体进行了CCS测序,尽管进行了广泛检测,包括短读长基因组测序,但这些三联体的病因仍无法解释。我们在每个三联体中鉴定了变异并创建了组装体,整体指标表明这些数据集比短读长数据提供的数据集更准确、更全面。在一个先证者中,我们鉴定出一个可能致病的(LP)由L1介导的插入,该插入导致外显子3重复,从而导致移码。在第二个先证者中,我们鉴定出多个大的结构变异,包括影响[具体基因1]和[具体基因2]的插入-易位,我们发现这些变异会破坏转录本水平。我们认为这种广泛的结构变异可能具有致病性。变异检测的广度和质量,以及在六个病因不明的NDDs先证者中的两个中发现具有临床和研究意义的变异,支持了长读长基因组测序可以大幅提高罕见病基因发现率这一假设。