Jensen Tanner D, Ni Bohan, Reuter Chloe M, Gorzynski John E, Fazal Sarah, Bonner Devon, Ungar Rachel A, Goddard Pagé C, Raja Archana, Ashley Euan A, Bernstein Jonathan A, Zuchner Stephan, Greicius Michael D, Montgomery Stephen B, Schatz Michael C, Wheeler Matthew T, Battle Alexis
Department of Genetics, Stanford University, Stanford, California 94305, USA.
Department of Computer Science, Johns Hopkins University, Baltimore, Maryland 21218, USA.
Genome Res. 2025 Apr 14;35(4):914-928. doi: 10.1101/gr.279323.124.
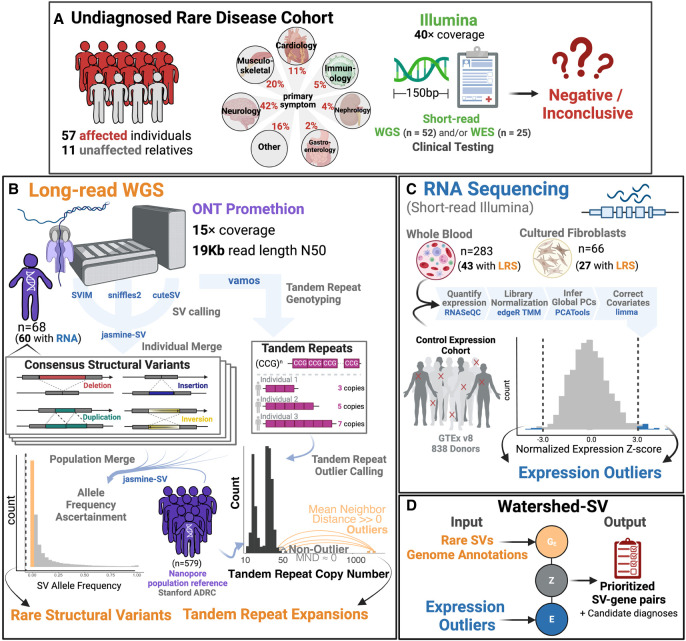
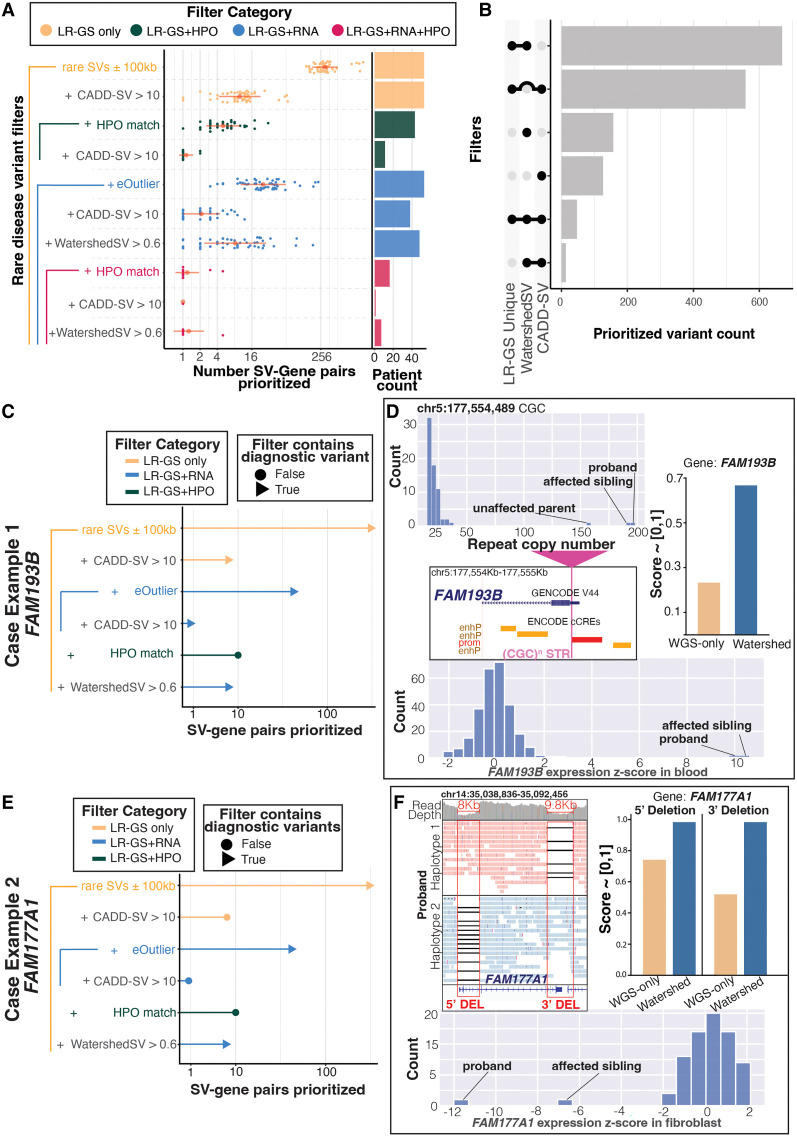
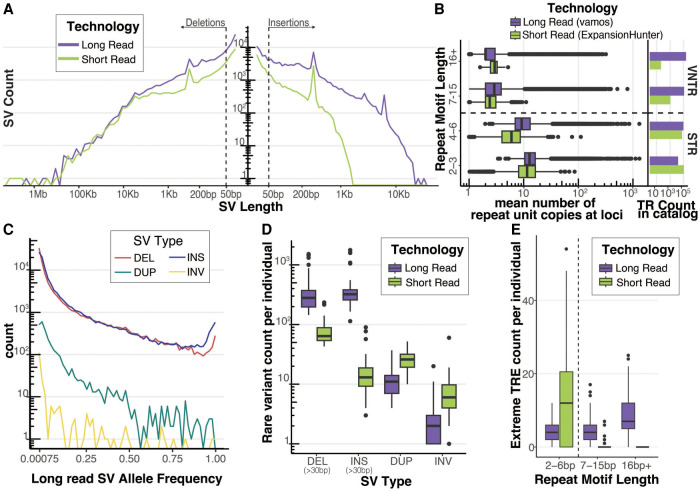
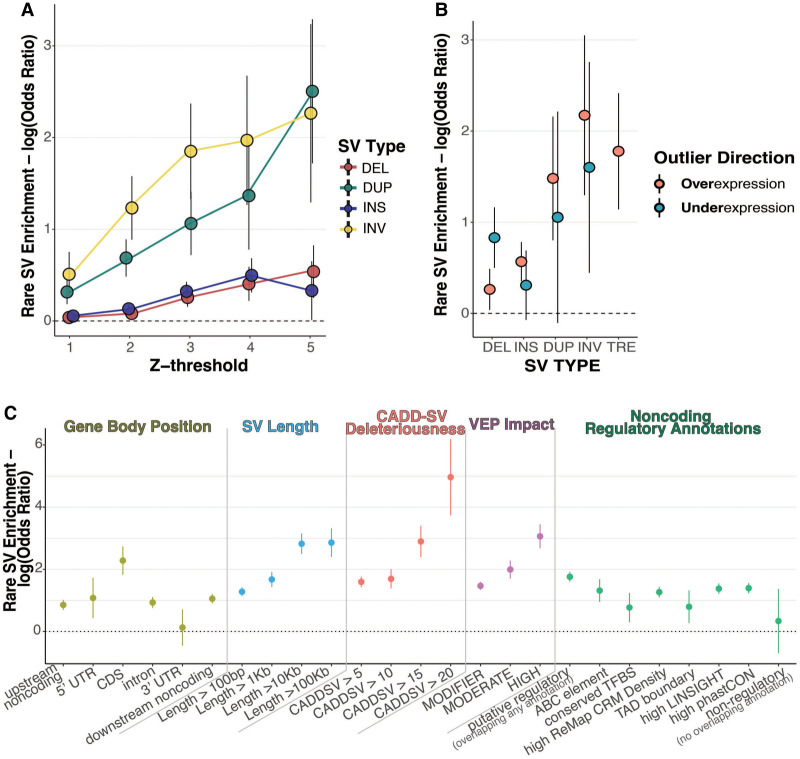
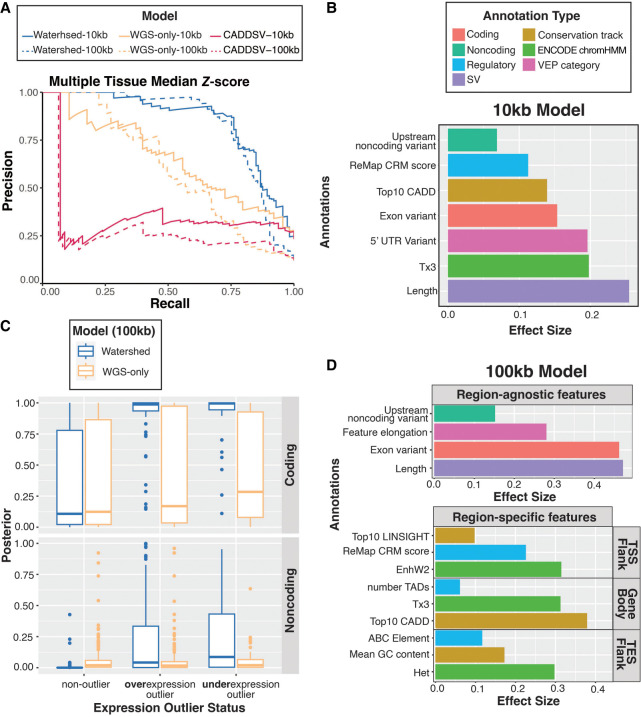
Rare structural variants (SVs)-insertions, deletions, and complex rearrangements-can cause Mendelian disease, yet they remain difficult to accurately detect and interpret. We sequenced and analyzed Oxford Nanopore Technologies long-read genomes of 68 individuals from the undiagnosed disease network (UDN) with no previously identified diagnostic mutations from short-read sequencing. Using our optimized SV detection pipelines and 571 control long-read genomes, we detected 716 long-read rare (MAF < 0.01) SV alleles per genome on average, achieving a 2.4× increase from short reads. To characterize the functional effects of rare SVs, we assessed their relationship with gene expression from blood or fibroblasts from the same individuals and found that rare SVs overlapping enhancers were enriched (LOR = 0.46) near expression outliers. We also evaluated tandem repeat expansions (TREs) and found 14 rare TREs per genome; notably, these TREs were also enriched near overexpression outliers. To prioritize candidate functional SVs, we developed Watershed-SV, a probabilistic model that integrates expression data with SV-specific genomic annotations, which significantly outperforms baseline models that do not incorporate expression data. Watershed-SV identified a median of eight high-confidence functional SVs per UDN genome. Notably, this included compound heterozygous deletions in shared by two siblings, which were likely causal for a rare neurodevelopmental disorder. Our observations demonstrate the promise of integrating long-read sequencing with gene expression toward improving the prioritization of functional SVs and TREs in rare disease patients.
罕见结构变异(SVs)——插入、缺失和复杂重排——可导致孟德尔疾病,但它们仍难以准确检测和解读。我们对来自未确诊疾病网络(UDN)的68名个体的牛津纳米孔技术长读长基因组进行了测序和分析,这些个体之前的短读长测序未发现诊断性突变。使用我们优化的SV检测流程和571个对照长读长基因组,我们平均每个基因组检测到716个长读长罕见(MAF < 0.01)SV等位基因,比短读长检测数量增加了2.4倍。为了表征罕见SVs的功能效应,我们评估了它们与来自同一受试者血液或成纤维细胞的基因表达之间的关系,发现与增强子重叠的罕见SVs在表达异常值附近富集(LOR = 0.46)。我们还评估了串联重复扩增(TREs),每个基因组发现14个罕见TREs;值得注意的是,这些TREs在过表达异常值附近也有富集。为了对候选功能SVs进行优先级排序,我们开发了Watershed-SV,这是一种将表达数据与SV特异性基因组注释整合的概率模型,其性能显著优于未纳入表达数据的基线模型。Watershed-SV在每个UDN基因组中鉴定出中位数为8个高置信度功能SVs。值得注意的是,这包括两个兄弟姐妹共有的复合杂合缺失,这可能是一种罕见神经发育障碍的病因。我们的观察结果表明,将长读长测序与基因表达相结合有望改善罕见病患者中功能SVs和TREs的优先级排序。