Bepari Asim Kumar, Reza Hasan Mahmud
Department of Pharmaceutical Sciences, North South University, Dhaka, Bangladesh.
PeerJ. 2021 Apr 13;9:e11261. doi: 10.7717/peerj.11261. eCollection 2021.
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has ravaged lives across the globe since December 2019, and new cases are still on the rise. Peoples' ongoing sufferings trigger scientists to develop safe and effective remedies to treat this deadly viral disease. While repurposing the existing FDA-approved drugs remains in the front line, exploring drug candidates from synthetic and natural compounds is also a viable alternative. This study employed a comprehensive computational approach to screen inhibitors for SARS-CoV-2 3CL-PRO (also known as the main protease), a prime molecular target to treat coronavirus diseases.
We performed 100 ns GROMACS molecular dynamics simulations of three high-resolution X-ray crystallographic structures of 3CL-PRO. We extracted frames at 10 ns intervals to mimic conformational diversities of the target protein in biological environments. We then used AutoDock Vina molecular docking to virtual screen the Sigma-Aldrich MyriaScreen Diversity Library II, a rich collection of 10,000 druglike small molecules with diverse chemotypes. Subsequently, we adopted in silico computation of physicochemical properties, pharmacokinetic parameters, and toxicity profiles. Finally, we analyzed hydrogen bonding and other protein-ligand interactions for the short-listed compounds.
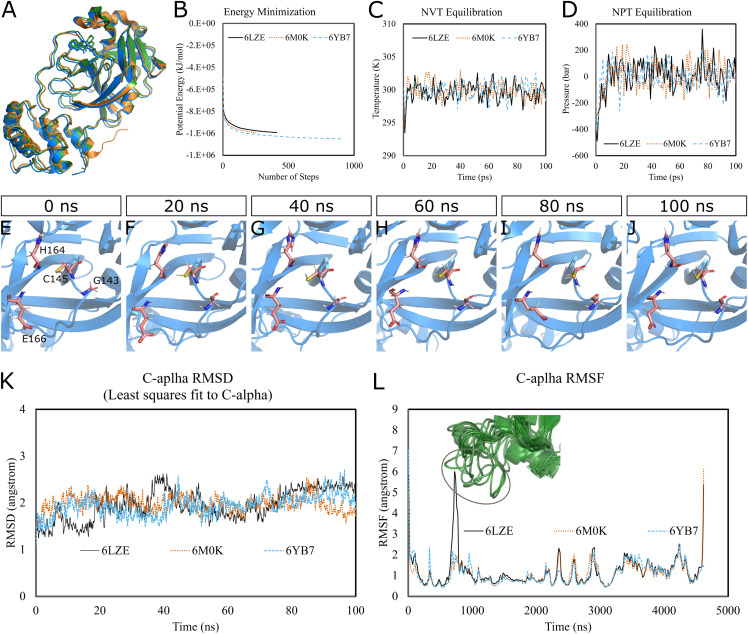
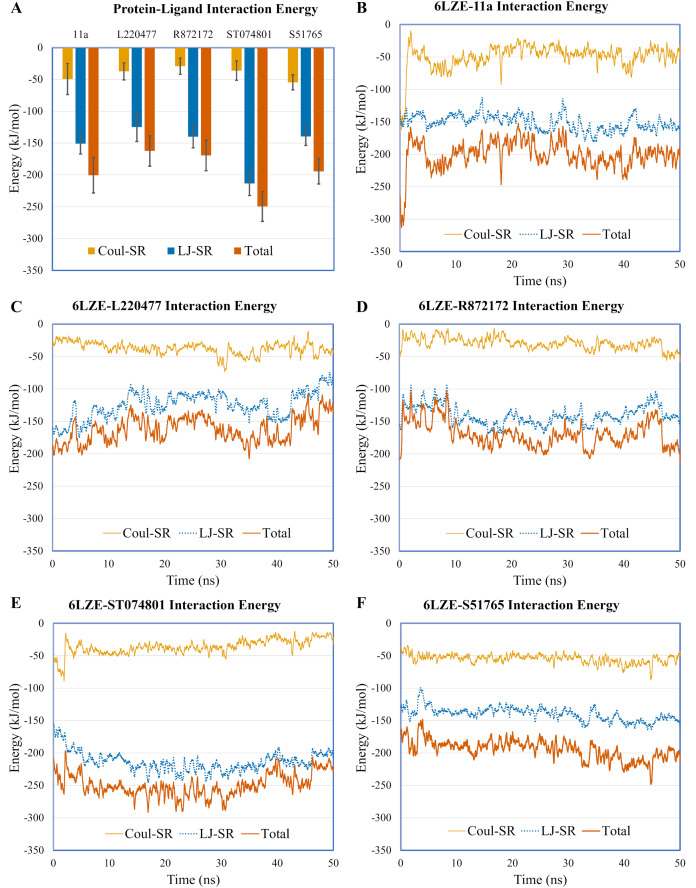
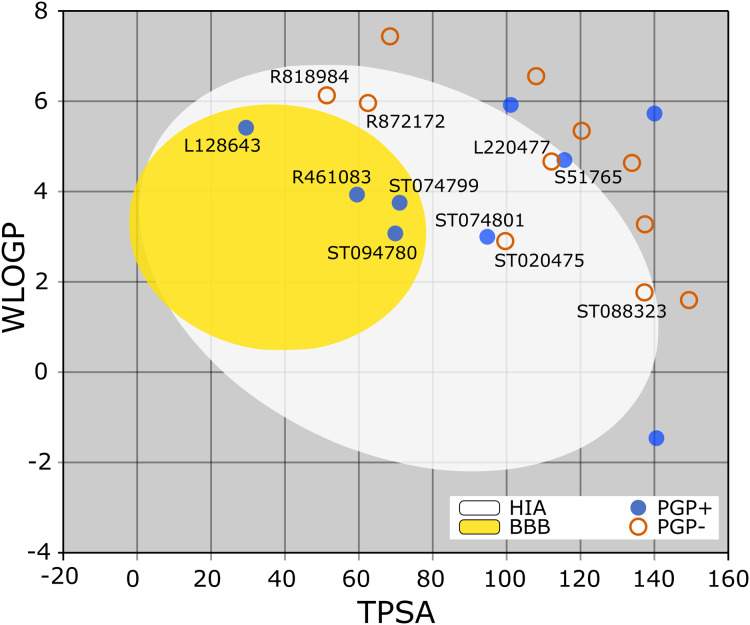
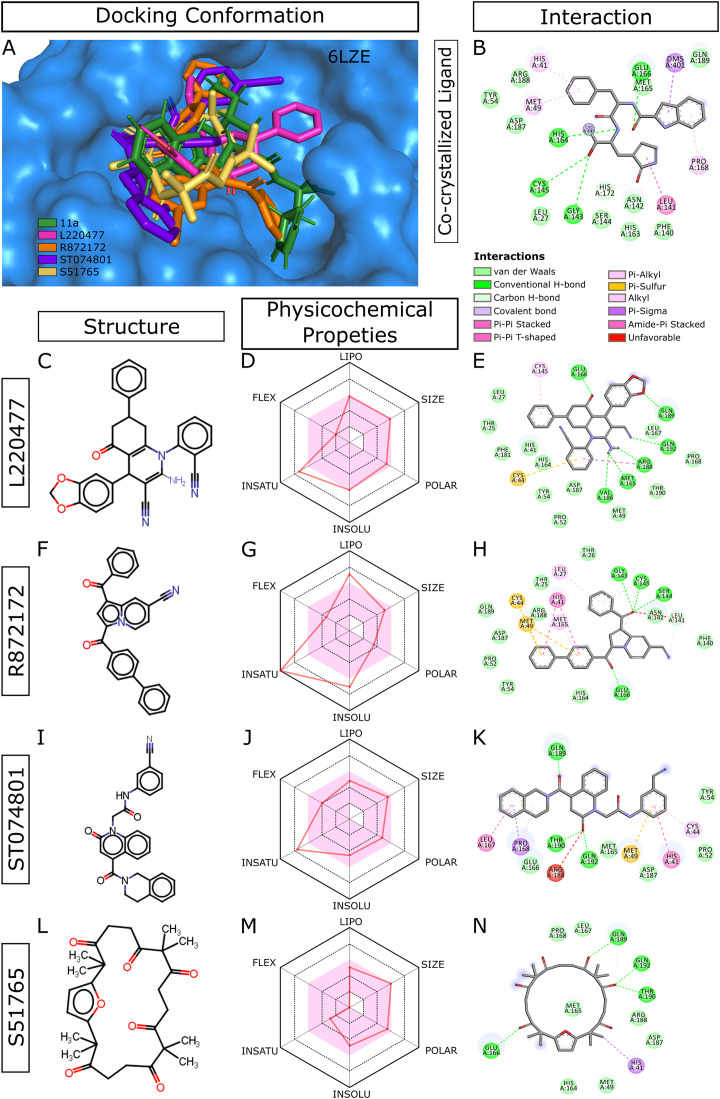
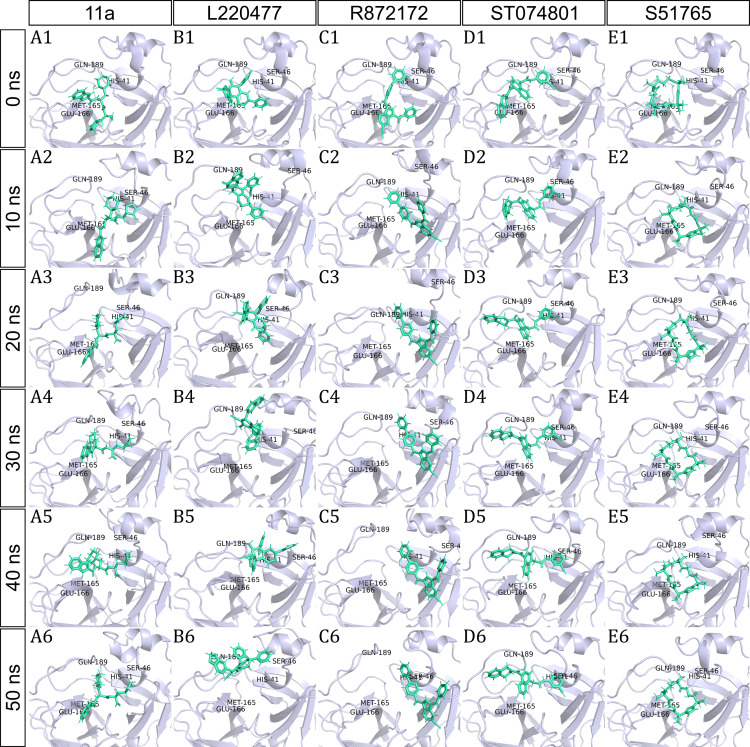
Over the 100 ns molecular dynamics simulations of 3CL-PRO's crystal structures, 6LZE, 6M0K, and 6YB7, showed overall integrity with mean Cα root-mean-square deviation (RMSD) of 1.96 (±0.35) Å, 1.98 (±0.21) Å, and 1.94 (±0.25) Å, respectively. Average root-mean-square fluctuation (RMSF) values were 1.21 ± 0.79 (6LZE), 1.12 ± 0.72 (6M0K), and 1.11 ± 0.60 (6YB7). After two phases of AutoDock Vina virtual screening of the MyriaScreen Diversity Library II, we prepared a list of the top 20 ligands. We selected four promising leads considering predicted oral bioavailability, druglikeness, and toxicity profiles. These compounds also demonstrated favorable protein-ligand interactions. We then employed 50-ns molecular dynamics simulations for the four selected molecules and the reference ligand 11a in the crystallographic structure 6LZE. Analysis of RMSF, RMSD, and hydrogen bonding along the simulation trajectories indicated that S51765 would form a more stable protein-ligand complexe with 3CL-PRO compared to other molecules. Insights into short-range Coulombic and Lennard-Jones potentials also revealed favorable binding of S51765 with 3CL-PRO.
We identified a potential lead for antiviral drug discovery against the SARS-CoV-2 main protease. Our results will aid global efforts to find safe and effective remedies for COVID-19.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病疫情自2019年12月以来已在全球肆虐,新病例仍在增加。人们持续遭受的痛苦促使科学家们研发安全有效的疗法来治疗这种致命的病毒性疾病。虽然重新利用美国食品药品监督管理局(FDA)批准的现有药物仍处于前沿,但从合成和天然化合物中探索候选药物也是一种可行的选择。本研究采用了一种全面的计算方法来筛选针对SARS-CoV-2 3CL蛋白酶(也称为主要蛋白酶)的抑制剂,这是治疗冠状病毒疾病的主要分子靶点。
我们对3CL蛋白酶的三个高分辨率X射线晶体结构进行了100纳秒的GROMACS分子动力学模拟。我们以10纳秒的间隔提取帧,以模拟目标蛋白在生物环境中的构象多样性。然后,我们使用AutoDock Vina分子对接对西格玛奥德里奇公司的MyriaScreen Diversity Library II进行虚拟筛选,该库包含10000种具有不同化学类型的类药物小分子。随后,我们对物理化学性质、药代动力学参数和毒性特征进行了计算机模拟计算。最后,我们分析了入围化合物的氢键和其他蛋白质-配体相互作用。
在对3CL蛋白酶晶体结构6LZE、6M0K和6YB7进行的100纳秒分子动力学模拟中,整体结构保持完整,平均Cα均方根偏差(RMSD)分别为1.96(±0.35)埃、1.98(±0.21)埃和1.94(±0.25)埃。平均均方根波动(RMSF)值分别为1.21±0.79(6LZE)、1.12±0.72(6M0K)和1.11±0.60(6YB7)。在对MyriaScreen Diversity Library II进行两阶段的AutoDock Vina虚拟筛选后,我们列出了前20种配体。考虑到预测的口服生物利用度、类药物性质和毒性特征,我们选择了四种有前景的先导化合物。这些化合物还表现出良好的蛋白质-配体相互作用。然后,我们对四个选定的分子和晶体结构6LZE中的参考配体11a进行了50纳秒的分子动力学模拟。对模拟轨迹上的RMSF、RMSD和氢键的分析表明,与其他分子相比,S51765与3CL蛋白酶形成的蛋白质-配体复合物更稳定。对短程库仑势和伦纳德-琼斯势的深入分析也揭示了S51765与3CL蛋白酶的有利结合。
我们确定了一种针对SARS-CoV-2主要蛋白酶的抗病毒药物发现的潜在先导化合物。我们的结果将有助于全球寻找治疗2019冠状病毒病的安全有效疗法的努力。